


 收藏
收藏 已收藏
已收藏作 者:梁梓宇(主治医师 南宁市第二人民医院消化内科)
指导者:姜海行(主任医师 广西医科大学第一附属医院消化内科)
淀粉样变是一组不同结构的蛋白质在不同组织的细胞外间质和血管壁沉积而引起的病变,主要累及心、肾、肝、脾、胃肠、肌肉、神经和皮肤等组织器官。肝脏受累者,即为肝淀粉样变,常为系统性淀粉样变的一部分。局灶性肝淀粉样变罕见。
1.发病机制
肝淀粉样变病因迄今尚未完全明了,一般认为与以下因素有关:①理化因素;②细菌毒素;③免疫因素;④遗传因素;⑤浆细胞病;⑥肿瘤性疾病和其他因素,如长期透析相关淀粉样变(β2-微球蛋白)等。淀粉样物质为一类结合黏多糖的蛋白质,遇碘时被染成褐色,加硫酸成蓝色,至少包含纤维蛋白、淀粉样P蛋白和黏多糖3种成分,95%是纤维蛋白。淀粉样物质常见有以下几种:①AL型蛋白;②AA型蛋白;③转甲状腺蛋白;④β-蛋白;⑤β2微球蛋白;⑥其他:如载脂蛋白A-1。
2.肝淀粉样变分类
根据疾病的获得方式、生化属性以及临床特点的不同,将淀粉样变分为原发性、继发性、遗传性等类型。
(1)原发性淀粉样变(或称AL型淀粉样变性):50%以上有肝淀粉样变。这是一种没有明确原发或共存疾病的临床综合征。淀粉样物质来源于免疫球蛋白轻链或轻链片段(AL型蛋白)。依据患者是否存在多发性骨髓瘤可分2类:无多发骨髓瘤和伴多发骨髓瘤。后者骨髓内可出现骨髓瘤细胞,血清及尿中出现M蛋白,有高钙血症,X线检查可见溶骨性病变,预后差。
(2)继发性淀粉样变(或称AA型淀粉样变):约25%有肝淀粉样变。这是一种与多种炎症性疾病相关的临床综合征。淀粉样A蛋白(AA蛋白)是由血清中的一种非免疫球蛋白的血清淀粉样蛋白A分解而成,由76个氨基酸组成的单链多肽。其与C反应蛋白和补体C3一样,是一种急性炎症反应物质。正常情况下,单核细胞的弹力蛋白酶能降解血清淀粉样蛋白A。继发性淀粉样变患者的单核细胞降解血清淀粉样蛋白A功能出现障碍,导致淀粉样蛋白A在组织中沉积。该病慢性发病,继发于慢性感染(如结核、慢性肺化脓性感染、慢性骨髓炎)、自身免疫疾病(如类风湿性关节炎、强直性脊柱炎、炎性肠病)、肿瘤(如恶性淋巴瘤、霍奇金病、甲状腺髓样癌等)、遗传性家族性疾病(如家族性地中海热等)、内分泌相关性疾病等。
(3)遗传性淀粉样变:以家族形式出现,是常染色体显性遗传病。常见淀粉样物质有转甲状腺蛋白、载脂蛋白A-1及纤维蛋白原等蛋白的突变体。约有10%肝淀粉样变。
(4)老年性淀粉样变性。
(5)局限性或肿瘤形成性淀粉样变:如透析相关淀粉样变。
3.临床表现
主要表现为肝大、上腹饱满、食欲缺乏,乏力、体重下降,水肿。少数患者出现严重肝大。其他表现还有肺受累而出现咳嗽或呼吸困难,皮肤紫癜性或丘疹性皮损,腕管综合征,充血性心力衰竭,自主神经病变,直立性低血压,胃肠道受累而出现消化道出血、腹泻或吸收不良,肾病综合征等。
4.诊断
(1)本病呈慢性病程,多发于中老年人。患者常因不明原因的疲乏、无力、体重下降等症状就诊。查体及辅助检查发现不明原因的肝大,伴随下列情形,可疑诊为肝淀粉样变:①原因不明的其他器官功能不全,如肾病综合征、肾功能不全、心功能不全等;②腕管综合征、外周神经感觉异常、蜡状皮肤病和巨舌症;③血、尿中出现M蛋白;④骨髓穿刺出现单克隆浆细胞。
(2)本病诊断依靠活检组织病理学检查。一般行腹壁脂肪、直肠黏膜、牙龈和受累组织器官活检。刚果红染色后在偏光镜下观察,淀粉样变表现为特异的双折射绿光。如果用改良的高锰酸钾预先处理,可使继发性淀粉样变失去对刚果红染色的亲和力,偏光显微镜下绿色折射消失,借此可区分原发性淀粉样变和继发性淀粉样变。
5.鉴别诊断
(1)与引起肝脾大的肝脏疾病鉴别
如急慢性肝炎、胆汁性肝硬化、慢性血吸虫肝病、肝豆状核变性、肝静脉阻塞综合征、肝癌等。
(2)与引起肝脾大的心脏疾病鉴别
如缩窄性心包炎、高血压心力衰竭等。
(3)与引起肝脾大的血液病鉴别
如急慢性粒细胞白血病、浆细胞病等。
6.治疗
治疗目的是祛除致淀粉样变性的病因,抑制淀粉样物质的合成,防止淀粉样物质进一步沉积,促进或加速已沉积的淀粉样物质的吸收。
(1)原发性淀粉样变治疗
美法仑+地塞米松(MD方案)用法为美法仑0.15mg/(kg.d)分两次口服,地塞米松40mg/d静脉滴注4天,每4周重复一个疗程。
(2)继发性淀粉样变治疗
治疗原发病;使用二甲亚砜、秋水仙碱。
(3)遗传性淀粉样变治疗
肝移植。
7.预后
本病预后较差,自然病程1~5年。原发性淀粉样变患者平均生存期为2年。有多发骨髓瘤患者预后最差,常1年内死亡。常见死因为继发感染和心力衰竭、肾衰竭。可能的死因有肝衰竭、消化道出血、呼吸衰竭。
1.患者基本情况
患者:男性,55岁。
入院时间:2016年2月24日。
主诉:反复右上腹痛3个月余,加重伴腹胀7天。
现病史:患者于3个月余前无明显诱因下出现右上腹痛,呈持续隐痛,疼痛程度轻,可忍受,无放射性,进餐后及活动后加重,伴有头晕、乏力,无恶心、呕吐,无胸闷、心悸,无咳嗽、咳痰,未进一步诊治。7天前患者上腹痛加重,性质同前,伴有轻度腹胀,与进食无明显关系,来我院就诊,门诊以“肝硬化、腹水”于2016年2月24日收住我科。
既往史:2015年3月,患者因“右胁不适”在本院门诊就诊,诊断为“肝硬化?胆囊炎,脾大”,具体治疗不详。2个月前,患者因右足外伤在当地医院治疗。患者在外地工作3年,接触化纤材料3年,余无特殊。
个人史:偶有吸烟、饮酒,有食鱼生史。否认伤寒、结核或其他传染病史;否认过敏史;否认滥用药物史;否认手术史,否认输血史。
家族史:否认高血压、糖尿病、慢性肾病等家族史,家族中无类似疾病患者。
2.入院查体
生命体征:T 37.1℃,P 87 次/min,R 20次 /min,BP 105/68mmHg。
查体:神清,慢性病容,巩膜轻度黄染,未见肝掌及蜘蛛痣。两肺呼吸音粗,未闻及干湿啰音。心律齐,未闻及杂音。腹部稍隆,未见腹壁静脉曲张及胃肠蠕动波。腹壁软,右上腹压痛,无反跳痛。肝于肋下6cm可及,质硬,边缘钝,未触及结节。脾于肋下4cm可触及,质地中等,边缘钝,无触痛。肝区叩击痛(+),肾脾区无叩击痛,移动性浊音可疑阳性,肠鸣音活跃。双下肢轻度凹陷性水肿。
3.入院辅助检查(2016年2月24日,本院)
血细胞分析(急):白细胞计数10.1×109/L,中性粒细胞计数7.4×109/L,红细胞计数4.26×1012/L,血红蛋白129g/L,血小板计数159×109/L。
肝功能:总胆红素31.0µmol/L,直接胆红素18.2µmol/L,白蛋白32.1g/L,谷氨酰转肽酶280U/L,碱性磷酸酶317U/L,腺苷脱氨酶44.4U/L,总胆汁酸18.66µmol/L。
肾功能:尿酸493µmol/L。
脂肪酶82.1U/L。
胃肠肿瘤相关检查:糖类抗原199(carbohydrate antigen 199,CA199)45.15U/mL,糖类抗原 125(CA125)260.80U/mL。
电解质、心肌酶、凝血无异常。
肝病自身抗体、输血前四项(乙肝病毒表面抗原、丙肝病毒抗体、HIV抗体、梅毒螺旋体抗体)均阴性。
无痛电子胃镜:①食管静脉曲张;②慢性非萎缩性胃炎;③胃体多发毛细血管扩张;④胃底胃窦多发黄色素瘤(图1)。
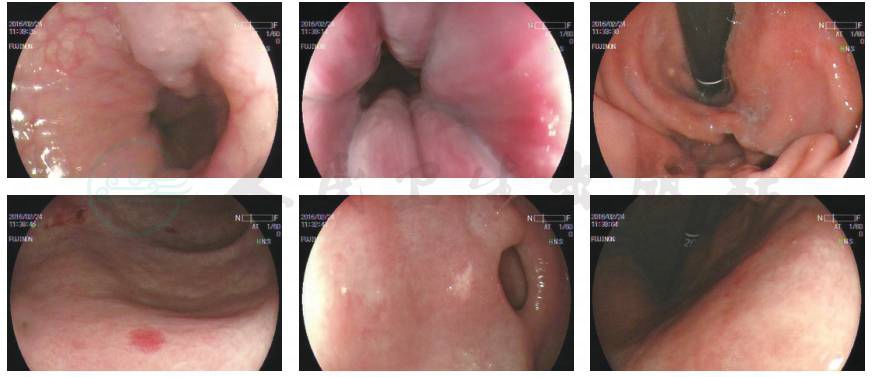
图1 无痛胃镜检查(2016年2月24日)
引自:主编:.消化系统疑难疾病诊疗思维及病例解析.第1版.ISBN:978-7-117-28669-5
病理学检查:①(胃底、胃窦)胃黄斑;②(胃窦)非萎缩性慢性胃窦炎,间质水肿,幽门螺杆菌(helicobacter pylori,Hp)(+)(图2)。
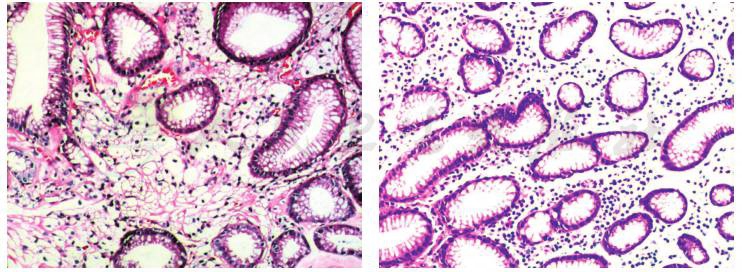
图2 胃镜病理(HE染色,100×)(2016年2月26日)
引自:主编:.消化系统疑难疾病诊疗思维及病例解析.第1版.ISBN:978-7-117-28669-5
上腹部CT平扫+增强+重建:①考虑肝硬化并侧支循环形成、少量腹水,建议定期复查;②脾大,脾包膜钙化;③胆囊多发结石并胆囊炎;④两肺下叶炎性病变并两侧胸腔少量积液;⑤心包少量积液(图3)。
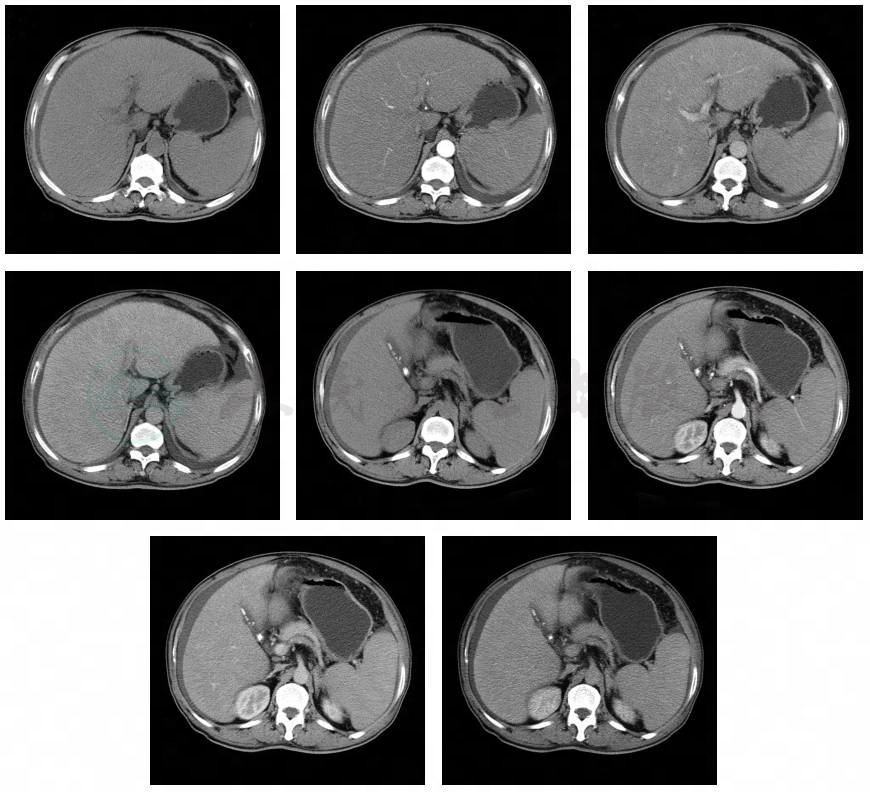
图3 上腹部增强CT(2016年2月25日)
引自:主编:.消化系统疑难疾病诊疗思维及病例解析.第1版.ISBN:978-7-117-28669-5
4.初步诊断思维过程
(1)入院时病情总结
①患者为中年男性,出现反复右上腹痛3个月余,逐渐加重,伴有腹胀;②体格检查有肝脾大,腹水征;③既往曾考虑诊断肝硬化,胃镜检查见门脉高压表现,腹部增强CT支持肝硬化诊断。
(2)入院时诊断思路
1)病史特点
患者为中年男性,临床表现为右上腹痛伴乏力、腹胀。2015年曾诊断肝硬化可能,胆囊炎、脾大。
2)查体
慢性病容,巩膜轻度黄染,右上腹压痛,肝脾可触及。
3)辅助检查
①肝功能稍异常,肝病自身抗体、输血前四项均阴性;②胃镜检查见食管静脉曲张;③上腹部CT平扫+增强+重建,考虑肝硬化并侧支循环形成、少量腹水,并见脾大,胆囊多发结石并胆囊炎,两肺下叶炎性病变并两侧胸腔少量积液。
(3)入院初步诊断
①肝脾大待查:肝硬化(病毒感染或中毒可能)?肝淀粉样变?巴德-吉亚利综合征?②胆囊多发结石并胆囊炎。③肺部感染。
5.后续检查及诊治
(1)2016年2月29日,行超声引导下肝脏病变穿刺活检术
用自动活检枪弹射18G内槽型切割针,取适宜角度经皮进针,在肝内组织取材3次,标本长度均为20mm。整个穿刺过程顺利,肝周无出血,患者无不适反应。
2016年3月1日凌晨1时,患者出现腹痛较前加重,伴腹胀明显,予腹部穿刺点处加压包扎,对症镇痛、利尿等处理,患者症状未缓解。
查体:腹部膨隆,移动性浊音呈阳性,予以诊断性腹腔穿刺术,抽出不凝固血性液体约3mL。
急查上腹部CT平扫(图4):①腹水较前增多。CT值约33HU,较前增高。肝第6段少量出血并肝周少量积血。②肝硬化并脾大、侧支循环形成。③脾包膜钙化。④胆囊多发结石并胆囊炎。⑤两肺下叶炎性病变并两侧胸腔少量积液。⑥心包积液,较前稍增多。
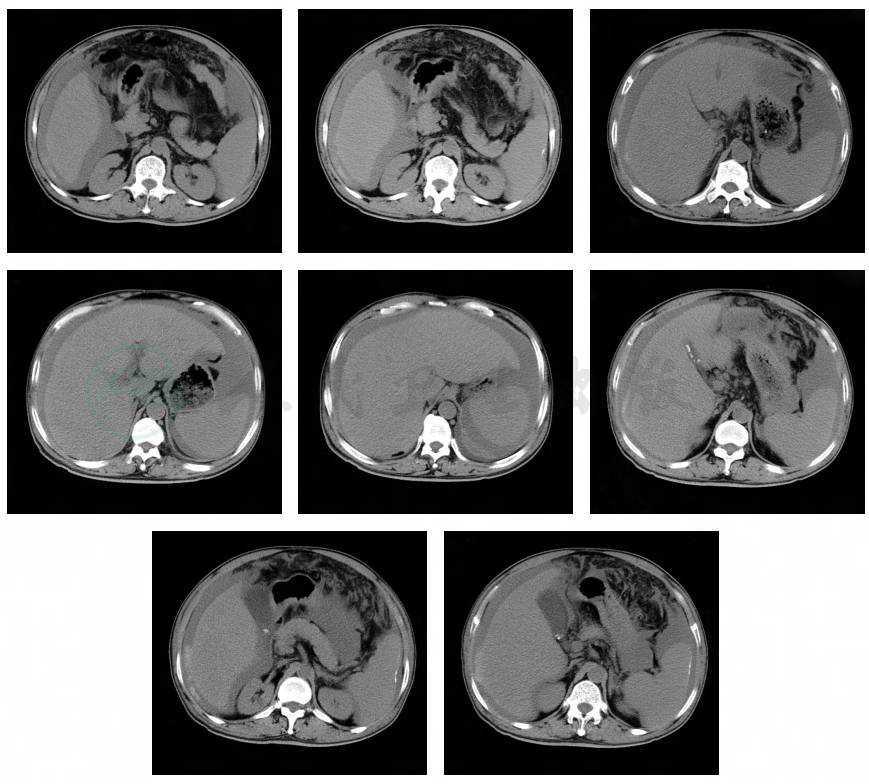
图4 上腹部平扫CT(2016年3月1日)
引自:主编:.消化系统疑难疾病诊疗思维及病例解析.第1版.ISBN:978-7-117-28669-5
超声造影提示腹腔内量造影剂外溢,考虑有活动性出血。
血细胞分析(急):白细胞计数13.4×109/L,中性粒细胞计数10.5×109/L,红细胞计数3.60×1012/L,血红蛋白108g/L,血细胞比容33.7%,血小板计数248×109/L。
考虑肝穿刺术后活动性出血,予止血、输液、配血等处理,转肝胆外科治疗。
(2)肝胆外科手术
术中见腹腔内血性液伴血块约6000mL。肝大,质硬,表面较多侧支循环,肝脏第6段见两处针眼大小活动性喷血。吸尽局部血液,用3-0普里灵线及薇乔肝针局部缝合,可见局部仍有渗血,立即给予局部填塞止血纱并压迫止血,后活动性出血处肝针缝合,予碘仿纱条局部填塞。经家属表示理解并签字同意后行局部碘仿纱条止血术,将10条碘仿纱条缝合连接后局部填塞止血,并留尾端至切口下缘戳孔引出。于右膈下及右肝下各放置一根引流管,管末端从切口下缘另戳小孔引出,固定引流管。
(3)肝穿刺活检病理
(我院)送检穿刺肝组织显示,大部分肝组织结构破坏,散在少量肿胀、变性的残存肝细胞,大片淀粉样物沉积,不排除肝淀粉样变性(图5)。标本院外会诊提示,未见明显的炎症反应和肿瘤,刚果红组织染色阳性(没有偏光显微镜证实),PAS染色阳性,根据形态学及组织化学染色,倾向于诊断肝淀粉样变。
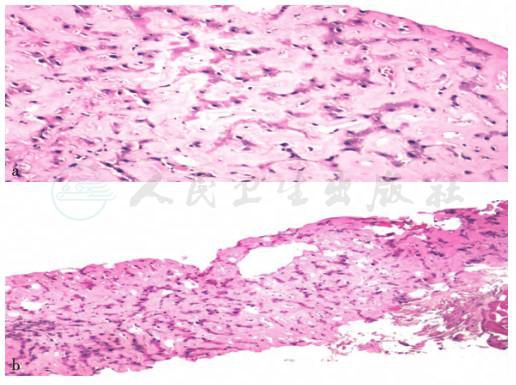
图5 肝穿刺病理(HE染色,100×)(2016年3月3日)
引自:主编:.消化系统疑难疾病诊疗思维及病例解析.第1版.ISBN:978-7-117-28669-5
6.最终诊疗思维过程
(1)最终诊断思路
肝淀粉样变呈慢性病程,多发于中老年人,患者常因肝大、上腹饱满、乏力、水肿等症状就诊。查体及辅助检查发现不明原因的肝大,伴随下列情形可疑诊为肝淀粉样变:①原因不明的其他器官功能不全,如肾病综合征、肾功能不全、心功能不全等;②腕管综合征、外周神经感觉异常、蜡状皮肤病和巨舌症;③血、尿中出现M蛋白;④骨髓穿刺出现单克隆浆细胞。
肝淀粉样变确诊主要依靠活检组织病理学检查,一般行腹壁脂肪、直肠黏膜、牙龈和受累组织器官活检。刚果红染色后在偏光镜下观察,淀粉样变表现为特异的双折射绿光。如果用改良的高锰酸钾预先处理,可使继发性淀粉样变失去对刚果红染色的亲和力,偏光显微镜下绿色折射消失,可借此区分原发性淀粉样变和继发性淀粉样变。
该病例患者本院及院外的病理结果都支持肝淀粉样变诊断。
(2)最终诊断
①肝淀粉样变、肝穿刺后出血、失血性休克、慢性肝衰竭、食管胃底静脉曲张;②胆囊结石并胆囊炎;③肺部感染。
(3)治疗方案
行肝修补术后,予输红细胞、血浆、冷沉淀、凝血酶原复合物、白蛋白,补充脂溶性维生素、抗感染等处理,患者引流管处仍间断出血,且凝血功能逐渐变差,胆红素水平逐渐上升,出现肝衰竭。4月13日,家属放弃治疗。
7.后续随访
出院后电话随访,患者腹腔引流管处仍间断出血,皮肤、巩膜黄染加重,1周后去世。
1.引起肝脾大的病因较多,除肝淀粉样变之外,需要与多种疾病鉴别:①引起肝脾大的肝病,如急慢性肝炎、胆汁性肝硬化、慢性血吸虫肝病、肝豆状核变性、肝静脉阻塞综合征、肝癌等;②引起肝脾大的心脏疾病,如缩窄性心包炎、高血压心力衰竭等;③引起肝脾大的血液病,如急慢性粒细胞白血病、浆细胞病等。
2.该病例影像学及临床表现支持肝硬化诊断,但肝硬化常见病因如乙型肝炎、丙型肝炎、肝病自身抗体等指标阴性,为明确病因肝穿刺活检很有必要。
3.该例患者肝穿刺前肝功能Child-Push评分为A级,穿刺后出血难以控制,肝功能损害加重。肝穿刺引起大出血并且难以止血的病例少见。
[1] Uchiyama K,Tsukada N,Miyazaki K,et al. Primary systemic AL amyloidosis with remarkable calcification in the spleen. Rinsho Ketsueki,2014,55(3):334-339.
[2] Chaulagain CP,Comenzo RL. New insights and modern treatment of AL amyloidosis.Current Hematologic Malignancy Reports,2013,8(4):291-298.
[3] Shin YM. Hepatic amyloidosis. Korean Journal of Hepatology,2011,17(1):80.
[4] Park MA,Mueller PS,Kyle RA,et al. Primary (AL)hepatic amyloidosis: clinical features and natural history in 98 patients. Medicine,2003,82(5):291-298.
[5] Loudin M,Childers R,Zivony A,et al. Liver failure secondary to light-chain amyloidosis.American Journal of Medicine,2016,129(4):e19-e20.
[6] Hydes TJ,Aspinall RJ. Subacute liver failure secondary to amyloid light-chain amyloidosis.Gastroenterology & Hepatology,2012,8(3):205.
[7] Lachmann HJ,Goodman HJ,Gilbertson JA,et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med,2007,356(23):2361-2371.
[8] Lachmann HJ,Booth DR,Booth SE,et al. Misdiagnosis of hereditary amyloidosis as AL(primary)amyloidosis. N Engl J Med,2002,346(23):1786.









