


 收藏
收藏 已收藏
已收藏男性,28岁。
发现血尿,蛋白尿,高血压1年。
1年前患者发现血压升高 150/100mmHg。尿检红细胞(+++),蛋白尿(++),血肌酐正常,予对症降压治疗,血压可控制在正常范围。因近3个月发现血肌酐升高就诊。
无手术、外伤史。从未做过尿检,没有接受过大的手术,没有接受过血制品的输注。未婚。否认酗酒、抽烟和应用违禁药物史。否认重金属及毒物接触史,无长期使用中药及镇痛药史。
主诉家族中无尿毒症患者,但是家族成员未查过尿。
血压150/80mmHg,脉搏71次/分,律齐。头颈部、心、肺、腹部查体未见阳性体征。双下肢未见水肿。未及系统和局部的神经系统异常。
24小时尿蛋白定量4.38g,血 ALB 36g/L,尿红细胞 80~100/HP,90%以上为变形红细胞,血肌酐 146μmol/L,血尿酸 500μmol/L。
慢性肾炎综合征 慢性肾脏病3期 肾性高血压
患者临床表现为典型的慢性肾炎综合征,24小时尿蛋白定量4.38g,血ALB 36g/L,尿红细胞 80~100/HP,90%以上为变形红细胞,血肌酐 146μmol/L,血尿酸 500μmol/L,血免疫球蛋白均正常,ANA及抗dsDNA抗体阴性。双肾大小正常。临床上未发现明显的继发因素。家族中无尿毒症患者。因此患者接受了肾穿刺活检以明确诊断。
肾活检病理:常规免疫荧光(-),光镜检查可见肾小球硬化,系膜增生,肾小管萎缩和肾间质纤维化。电镜:可见肾小球基底膜弥漫分层,撕裂状改变,伴节段性基底膜变薄 (图1)。肾活检提示患者有特殊的基底膜病变,于是我们进一步检查了肾组织和皮肤Ⅳ型交原染色,肾活检组织免疫荧光检查示Ⅳ型胶原α3/α4链阴性,α5(Ⅳ)链除肾小球囊和部分远曲小管基底膜阳性,在GBM呈阴性。皮肤基底膜(EBM)Ⅳ型胶原α5链染色为阳性(图2)。提示Ⅳ型胶原α5链正常表达。Ⅳ型胶原α3/α4链表达异常。补充了听力及眼睛的检查,发现双耳中度神经性耳聋,右晶体前凸,左前囊下浑浊。综合上述检查,考虑为Alport综合征。
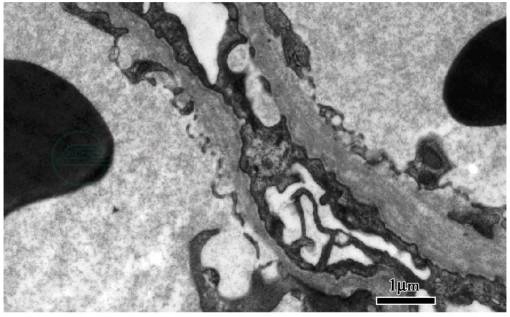
图1 Alport综合征:肾小球基底膜弥漫分层,撕裂状改变(电镜×10万)
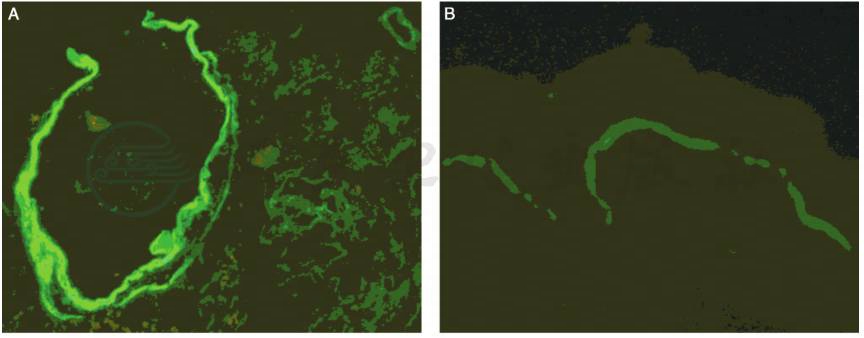
图2 Alport综合征:基底膜Ⅳ胶原α链免疫荧光染色(×400)。A:抗α5(Ⅳ)NC1抗体在包氏囊和部分TBM呈连续线状沉积,在GBM呈阴性反应;B:抗α5(Ⅳ)NC1抗体在EBM呈弱的连续状沉积
因为家族中无明确的尿毒症及肾脏病史,与常见的Alport综合征家族中多个肾衰竭及男重女轻的现象不符,我们进一步筛查了其他家系成员 (图3,表1)。发现患者的父母为表亲,其父母均表现为血尿蛋白尿,家族中有其他的成员有血尿蛋白尿,但是血肌酐均正常。经过基因检测,患者COL4A3第42外显子存在一个错义突变,即第3725位的鸟嘌呤突变为腺嘌呤(G3725A),导致Ⅳ型胶原α3链第1242位甘氨酸突变为天冬氨酸(G1242D)。
表1 患者家系筛查结果
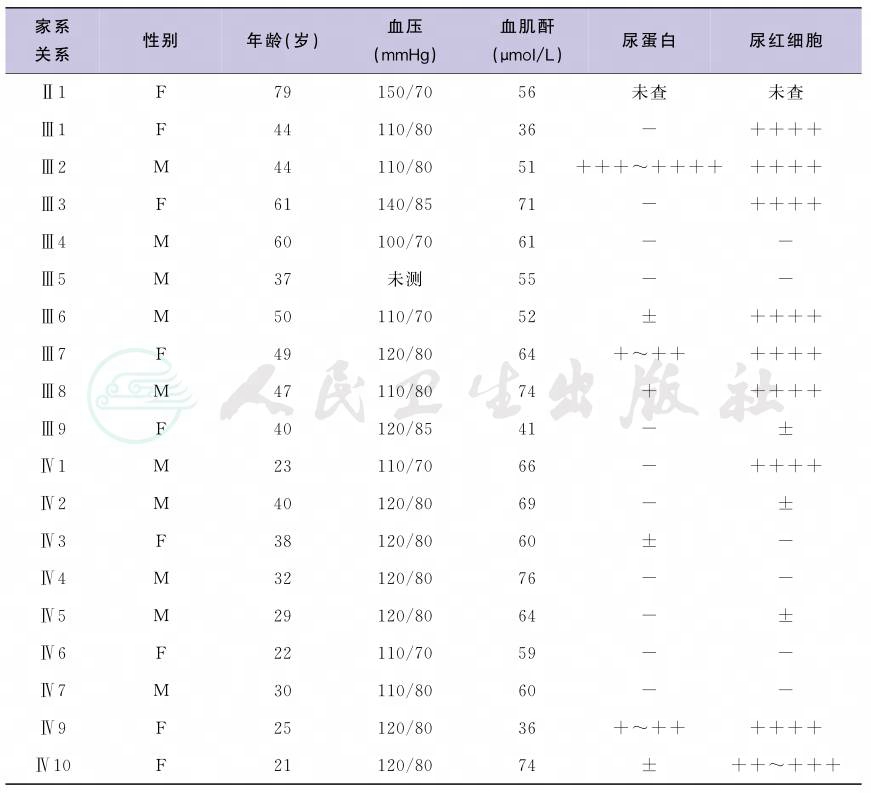
家系 关系 |
性别 |
年龄(岁) |
血压 (mmHg) |
血肌酐 (μmol/L) |
尿蛋白 |
尿红细胞 |
Ⅱ1 F 79 150/70 56 未查 未查 Ⅲ1 F 44 110/80 36 - ++++ Ⅲ2 M 44 110/80 51 +++~++++ ++++ Ⅲ3 F 61 140/85 71 - ++++ Ⅲ4 M 60 100/70 61 - - Ⅲ5 M 37 未测 55 - - Ⅲ6 M 50 110/70 52 ± ++++ Ⅲ7 F 49 120/80 64 +~++ ++++ Ⅲ8 M 47 110/80 74 + ++++ Ⅲ9 F 40 120/85 41 - ± Ⅳ1 M 23 110/70 66 - ++++ Ⅳ2 M 40 120/80 69 - ± Ⅳ3 F 38 120/80 60 ± - Ⅳ4 M 32 120/80 76 - - Ⅳ5 M 29 120/80 64 - ± Ⅳ6 F 22 110/70 59 - - Ⅳ7 M 30 110/80 60 - - Ⅳ9 F 25 120/80 36 +~++ ++++ Ⅳ10 F 21 120/80 74 ± ++~+++ |
||||||
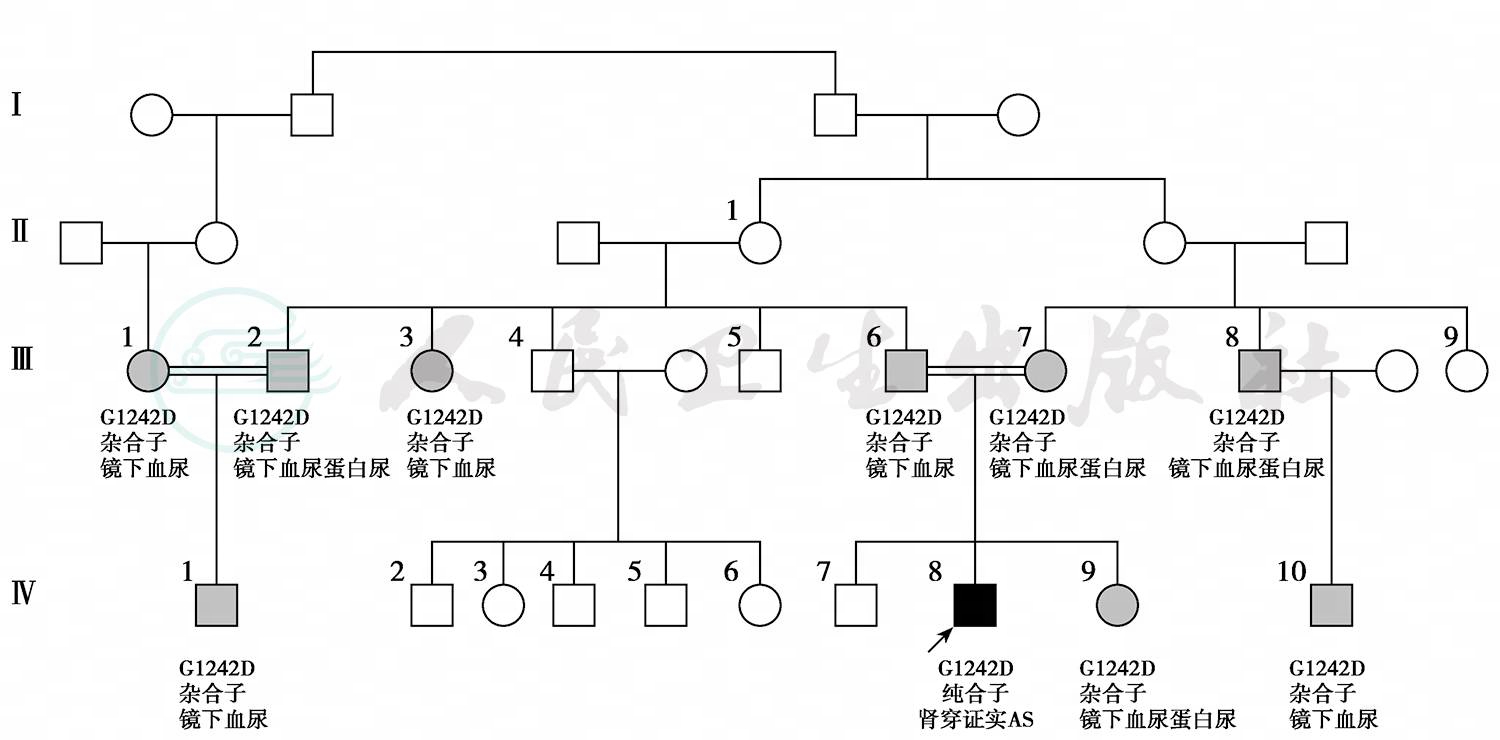
图3 患者家系图。圆圈代表女性,正方形代表男性,黑色为纯合子,灰色为杂合子
常染色体隐性遗传Alport综合征
Alport综合征(AS)是以持续性或反复性血尿、蛋白尿、进行性肾功能减退,并可合并高音区感音神经性耳聋和眼部异常等肾外改变为主要临床表现的一种遗传性疾病。其特征性病理改变为肾小球基底膜呈极不规则外观,基底膜弥漫性增厚或增厚与变薄相间,致密层劈裂、分层、篮网状改变。AS是一种遗传异质性疾病,AS的发生与基底膜主要成分Ⅳ型胶原α3~α6链编码基因COL4A3-COL4A6突变有关。其中大约 85%是由于COL4A5和COL4A6突变引起的X-连锁显性遗传型;还有大约15%患者是由位于2号染色体上编码Ⅳ型α3或α4链的COL4A3或CO4A4突变引起的常染色体隐性AS。
常染色体隐性遗传的AS由于病例数相对较少,患者的家族史常不明显(部分患者的父母有近亲婚配史),男性和女性均可发病,无性别差异,患者父母常不表现严重的临床症状(可以完全没有临床症状,或仅有单纯血尿),即使患者肾活检或其他肾外表现符合AS的诊断,有时对其是否遗传或遗传方式也很难明确判断。因此,建立和完善的常染色体隐性遗传AS基因诊断的方法,对患者进行基因突变分析,无疑对患者及其家系成员的遗传咨询、优生优育有着重要的指导意义。
患者家系中的致病性突变为COL4A3基因42外显子的第3725位核苷酸的鸟嘌呤突变为腺嘌呤,为错义突变,引起第1242位甘氨酸突变为天冬氨酸(G1242D)。这个错义突变在家系成员中随疾病分布,先证者为纯合突变,临床上表现为典型的Alport综合征,于30岁发展至慢性肾衰竭失代偿期,并伴有感音神经性耳聋及眼部典型病变,家族史调查显示父母双亲是近亲婚配。患者的肾活检组织及表皮组织的免疫荧光具有特征性的常染色体α3、4链表达异常表现:GBM 上α3、α4、α5(Ⅳ)链的同时缺失而 TBM、肾小球囊和皮肤基底膜(EBM)上α5(Ⅳ)链分布正常。而包括其父母在内的其他家系呈杂合状态(基因突变携带者)的成员,临床表现仅为镜下血尿,偶有蛋白尿,肾功能检查均显示正常;而非基因携带者则尿检正常。本家系中具有纯合的G1242D突变的先证者临床表型最重,而具有杂合的G1242D基因突变的患者临床表型较轻,提示基因剂量效应可能影响了该家系患者的临床表型。
1.Alport综合征(AS)是以持续性或反复性血尿、蛋白尿、进行性肾功能减退,并可合并高音区感音神经性耳聋和眼部异常等肾外改变为主要临床表现的一种遗传性疾病。其特征性的病理改变为肾小球基底膜呈极不规则外观,基底膜弥漫性增厚或增厚与变薄相间,致密层劈裂、分层、篮网状改变。
2.AS大约85%是由于COL4A5和COL4A6突变引起的X-连锁显性遗传型;还有大约15%患者是由位于2号染色体上编码Ⅳ型α3或α4链的COL4A3或CO4A4突变引起的常染色体隐性AS。
3.常染色体隐性遗传的AS由于病例数相对较少,患者家族史常不明显(部分患者的父母有近亲婚配史),发病无性别差异,患者父母常不表现严重的临床症状(可以完全没有临床症状,或仅有单纯血尿),即使患者肾活检或其他肾外表现符合AS的诊断,有时对其是否遗传或遗传方式也很难明确判断。
(侯 平 陈育青 张 宏)
1.Gubler MC,Knebelmann B,Beziau A,et al.Autosomal recessive Alport’s syndrome:immunohistochemical study of type Ⅳ collagen chain distribution.Kidney Int,1995,47(7):1142-1147.
2.Hou P,Chen Y,Ding J,et al.A novel mutation of COL4A3 presents a different contribution to Alport syndrome and thin basement membrane nephropathy.Am J Nephrol,2007,27(5):538-544.
3.Boye E,Mollet G,Forestier L,et al.C:Determination of the genomic structure of theCOL4A4 gene and of novel mutations causing autosomal recessive Alport’s syndrome.Am J Hum Genet,1998,63(5):1329-1340.
4.Zhang Y,Wang F,Ding J,et al.Genotype-phenotype correlations in 17 Chinese patients with autosomal recessive Alport syndrome.Am J Med Genet A,2012,158A(9):2188-2193.
5.Longo I,Scala E,Mari F,et al.Autosomal recessive Alport syndrome:an in-depth clinical and molecular analysis of five families.Nephrol Dial Transplant,2006,21(3):665-671.









