


 收藏
收藏 已收藏
已收藏男性,23岁。
发现血小板减少5年、蛋白尿1年。
患者5年前劳累后出现鼻出血、皮下出血点、神志障碍、发热;外院发现贫血和血小板减少,SCr升高(具体不详)。予血浆、糖皮质激素及丙种球蛋白后,症状渐缓解,SCr降至正常。后患者不规律服用激素及达那唑,多于激素减量或停药后出现头痛、头晕、口周及四肢麻木,伴血小板下降,当地医院予“激素”及血浆输注后改善。2年前停用激素。1年前尿检:蛋白(++),血压升高,予降压治疗。11天前外院实验室检查:SCr 272μmol/L;Hb 93g/L,PLT 21×109/L;为进一步诊治入院。患者发病来无光过敏、口腔溃疡或关节肿痛。
无特殊。
T 37.2℃,BP 160/100mmHg,HR 70次/分。库欣面容。心肺查未见异常。腹壁可见紫纹,余未见异常。双下肢不肿。
Hb 103g/L,Ret升高,PLT 340×109/L→110×109/L;非结合胆红素 25μmol/L 升高;LDH 266IU/L↑;外周血涂片可见破碎RBC 1%;Coombs试验(-);骨髓穿刺:巨核细胞增多,余未见异常。凝血功能正常。尿检:蛋白(++),定量 3.33g/d,RBC 5~7/HP;ALB 37.6g/L,SCr 296μmol/L。IgG 6.46g/L↓,IgA和IgM正常;C3 0.53g/L ↓,C4正常。ANA 1∶100,抗 ds-DNA和抗ENA谱(-),ACL(-)。PAIgG 38.00% ↑。血清ADAMTS-13活性:24%↓(正常值70%~80%),抗ADAMTS-13抗体(+)(IgG型)。血清H因子浓度正常,抗H因子抗体(-)。
贫血 血小板减少 肾功能不全原因待查
继发性高血压
患者青年男性,慢性病程。主要表现为PLT减少、贫血、中枢神经系统症状、发热、肾功能不全。需考虑以下情况:
(1)系统性红斑狼疮(SLE):患者多系统受累,包括ANA在内的多种自身抗体阳性,因此SLE不除外。
(2)血栓性微血管病(TMA):患者贫血为微血管病性溶血性贫血(MAHA),伴血小板减少、中枢神经系统和肾脏受累,凝血正常,需考虑此诊断。TMA倾向于根据病因分类,结合患者血清ADAMTS-13活性明显下降及抗ADAMTS-13抗体阳性,考虑为获得性血栓性血小板减少性紫癜(TTP)。需注意的是,SLE也可合并多种自身抗体,包括抗ADAMTS-13抗体。
(3)患者PAIgG阳性,考虑合并了自身免疫性血小板减少性紫癜。
下一步的肾穿刺活检对于明确TMA诊断及是否合并其他肾脏疾病非常重要。
肾穿刺活检(入院后第 3天)病理结果:免疫荧光:8个肾小球,IgG(+),IgM(+),C3(+),余(-),节段性系膜区颗粒样沉积。光镜:35个肾小球,肾小球系膜细胞和基质轻度弥漫增生,局灶节段中度加重伴内皮细胞增生,基底膜弥漫不规则增厚,节段性假双轨征形成,伴缺血皱缩,其中5个细胞纤维性、6个纤维性新月体形成;肾小管上皮空泡及颗粒变性,多灶状萎缩伴蛋白管型;肾间质多灶状淋巴和单核细胞浸润伴纤维化;小动脉管壁增厚,内膜葱皮状增生,管腔狭窄(图1)。
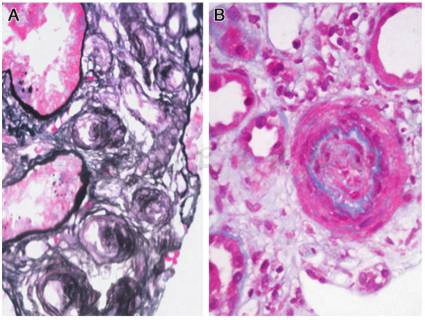
图1 血栓性血小板减少性紫癜:A,肾小叶间动脉内膜葱皮样增生,管腔狭窄(PASM×400);B,肾小叶间动脉内膜黏液变性,管腔狭窄(Masson×400)
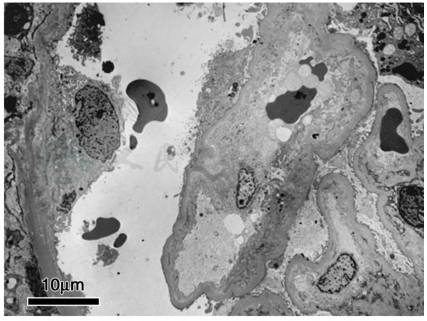
图2 血栓性血小板减少性紫癜:肾小球基底膜内疏松层增厚(电镜×5 000)
因经济原因,患者未接受血浆置换。遂予甲泼尼龙500mg×3天冲击治疗后改为泼尼松 60mg/d+环磷酰胺 50mg/d治疗,2周后 PLT 234×109/L,SCr 243μmol/L,LDH 132IU/L,ADAMTS-13活性升高至76%。
获得性血栓性血小板减少性紫癜 TMA肾损害伴部分新月体形成 慢性肾脏病4期
免疫性血小板减少性紫癜
本病例提供了一个典型的获得性TTP的范例。TTP最早由Moschowitz于1925年报道。1966年Amorosi等总结出TTP临床表现的五联征(PLT减少、MAHA、中枢神经系统异常、肾功能不全和发热),但之后的观察发现表现完整五联征者仅占5%。本例患者为典型的TTP五联征起病,同时其肾脏病理表现也符合典型TMA肾损害。
TTP又分为家族性和获得性。发病机制上,前者多为编码vWF剪切酶(ADAMTS-13)基因突变导致ADAMTS-13严重缺陷,后者多因抗ADAMTS-13抗体(多为IgG型)所致。ADAMTS-13是一种金属蛋白酶,存在于血浆和内皮细胞表面。vWF由内皮细胞分泌,在血浆中可形成超大分子的多聚体。正常情况下,超大分子的vWF可以被ADAMTS-13快速降解成小分子,否则将导致血小板聚集、血栓形成。
本例患者血清抗ADAMTS-13抗体阳性伴ADAMTS-13活性下降,最终确诊为获得性TTP,经强化免疫抑制疗法病情明显改善。国外指南推荐,TTP治疗首选血浆置换,家族性TTP可使用血浆输注;新发或复发获得性TTP可先激素冲击、后加用口服激素;严重蛋白酶缺乏者可加用免疫抑制剂;血浆置换无效或复发TTP可考虑使用抗CD20单抗治疗。另外,最新的研究显示,Caplacizumab(一种针对vWF分子A1区域的单克隆抗体)可以特异性阻断vWF分子与血小板Ib位点的结合,从而有效治疗TTP的患者,其应用前景值得关注。
在血浆疗法用于治疗TTP前,TTP死亡率达90%。血浆置换组患者,死亡率可降至22%;血浆输注治疗组患者,死亡率为37%。因此尽早诊断、尽早治疗可以明显改善患者预后。目前推荐,出现不能用其他原因解释的血小板减少和MAHA即可开始血浆置换治疗。
1.TTP典型临床表现为五联征,但出现概率低。
2.TTP分为家族性和获得性,前者多见于 ADAMTS-13基因突变,后者多见于针对ADAMTS-13的自身免疫。
3.TTP治疗首选血浆置换;获得性TTP可加用免疫抑制疗法。
(于 峰 陈 旻)
1.Moschcowitz E.An acute febrile pleiochromic anemia with hyaline thrombosis of the terminal arterioles and capillaries:an undescribed disease.1925.Mt Sinai J Med,2003,70(5):352-355.
2.George JN.How I treat patients with thrombotic thrombocytopenic purpura:2010.Blood,2010,116(20):4060-4069.
3.Rock GA,Shumak KH,Buskard NA,et al.Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura.Canadian Apheresis Study Group.N Engl J Med,1991,325(6):393-397.
4.Balduini CL,Gugliotta L,Luppi M,et al.High versus standard dose methylprednisolone in the acute phase of idiopathic thrombotic thrombocytopenic purpura:a randomized study.Ann Hematol,2010,89(6):591-596.
5.PeyvandiF,Scully M,Kremer Hovinga JA,et al.Caplacizumab for Acquired Thrombotic Thrombocytopenic Purpura.N Engl J Med,2016,374:511-522.









