


 收藏
收藏 已收藏
已收藏男性,71岁。
间断双下肢水肿、血肌酐升高4个月。
患者4个月前无诱因出现双下肢水肿,当地医院查尿常规未见异常,SCr 270μmol/L,曾予地塞米松和降压治疗,3个月前SCr 96μmol/L。2个月前患者服用“保健品”后出现皮疹,伴水肿、尿量减少,当地血常规:WBC 12.1×109/L,嗜酸性粒细胞 7.1%,Hb 89g/L,PLT 127×109/L;尿常规:蛋白(+++),潜血(+++);SCr 507μmol/L;骨穿提示继发性贫血;予利尿等对症治疗,SCr恢复至正常上限。10天前“受凉”后水肿再次加重,尿量明显减少,并逐渐出现意识障碍。3天前查SCr 1 200μmol/L,Hb 50g/L,PLT 30×109/L。予血液透析及输血等治疗,意识转清,持续无尿,为进一步诊治入我院。
“类风湿关节炎”5年,余无特殊。
T 36.5℃,HR 80次/分,BP 140/90mmHg。神志清楚,双手关节变形。肺、心和腹部查体无明显异常。双下肢轻度水肿。
血液系统:血常规 WBC 9.58×109/L,Hb 116g/L→89g/L,PLT 246×109/L→159×109/L,网织红细胞5%(升高);外周血涂片可见破碎 RBC 2%;间接胆红素 24.5μmol/L(升高);LDH 442U/L↑;凝血:D-二聚体 4.84mg/L↑,FDP 8.7mg/L↑,PT、APTT 和 FIB 均正常。
肾脏:尿检:蛋白(+++);RBC 15~30/HP,变形;WBC 10~18/HP,尿培养(-);NAG 酶 363U/L↑,α1微球蛋白 664mg/L↑。生化:ALB 33.9g/L,SCr 627μmol/L。
免疫方面:IgG 24.10g/L↑,IgA 和 IgM正常;C3 0.06g/L↓,C4 0.09g/L↓;Coombs试验(-);RF 512U/ml↑,抗CCP抗体>200RU/ml↑;双手X线:符合类风湿关节炎表现;ANA、抗 ds-DNA 和抗 ENA谱均(-);ACL(-);H 因子浓度 21.2μg/ml↓↓,抗 H 因子抗体(+)(IgG型);血清ADAMTS-13活性正常;ANCA和抗GBM均(-)。
其他:粪便培养(-)。B超:双肾大小正常,双肾实质回声偏强。
急性肾损伤原因待查
患者老年男性,临床主要表现为急性肾损伤(AKI),伴贫血、一过性PLT减少及神志障碍。诊断需考虑:
(1)血栓性微血管病(TMA)
患者存在微血管病性溶血性贫血(外周血破碎RBC增多,间接胆红素及LDH水平升高),伴PLT减少、ARF及中枢神经系统受累,有血栓形成的可能(D-二聚体和FDP水平升高),因此考虑TMA的诊断。TMA根据病因可分为多种,结合患者血清C3水平显著下降、H因子水平显著低于正常、抗H因子抗体阳性,分类应考虑为非典型溶血尿毒症综合征(aHUS)。
(2)急性肾小管间质性肾病(ATIN)
患者用药后出现皮疹、血 EOS升高,伴 ARF、无菌性白细胞尿、尿NAG和α1微球蛋白水平显著升高,因此考虑存在ATIN。但ATIN不能解释患者的变形血尿、神志障碍及PLT减少。
(3)自身免疫性疾病
患者存在类风湿关节炎,应考虑是否存在相关肾损害。
穿刺活检对于明确诊断非常重要。患者入院后第12天行肾活检,病理结果:免疫荧光:IgM(+),余(-),系膜区颗粒样沉积。光镜:可见 15个肾小球,1个球性硬化,其余全部缺血皱缩(图1);
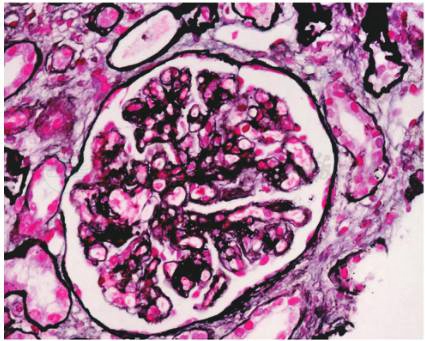
图1 血栓性微血管病:肾小球基底膜缺血皱缩。(PASM+Masson×400)
肾小管刷毛缘脱落,管腔扩张,灶状萎缩及蛋白管型;肾间质弥漫水肿,灶状淋巴和单核细胞浸润;小动脉管壁增厚,内膜葱皮状增生,管腔狭窄(图2);
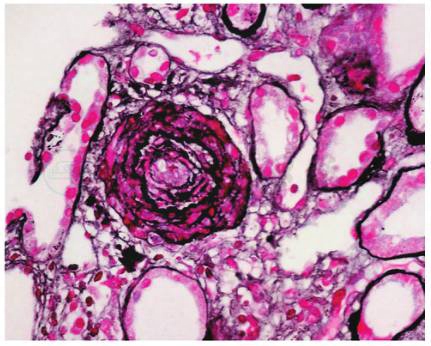
图2 血栓性微血管病:小动脉管壁增厚,内膜葱皮状增生,管腔狭窄。(PAMS+Masson×200)
符合 TMA伴ATIN肾损伤。电镜:肾小球系膜细胞和基质轻度增生,基底膜内疏松层增宽伴颗粒状结构分布,上皮足突大部分融合,肾小管萎缩,肾间质淋巴单核细胞浸润伴胶原纤维增生,符合光镜诊断(图3)。

图3 血栓性微血管病:肾小球基底膜内疏松增厚(电镜×5 000)
入院后予患者降压、血液透析及对症支持治疗。肾活检病理结果明确诊断后予血浆置换及甲泼尼龙40mg,每日1次治疗;随访1年,患者Hb和PLT维持正常,LDH渐正常,IgG下降,C3渐上升;抗H因子抗体转阴。但肾功能一直未恢复,予规律血液透析治疗。
非典型溶血尿毒综合征 TMA伴急性肾小管间质肾病肾损伤 急性肾损伤→慢性肾脏病5D期
类风湿关节炎
本例患者表现为ARF,最终确诊为TMA合并ATIN肾损伤。患者ATIN可能与药物及缺血相关;而TMA则是由于存在抗H因子抗体所致的不典型 HUS(aHUS)。
HUS是最经典的TMA之一,可分为d+HUS(由产志贺毒素的肠道细菌感染引起)及d-HUS(aHUS)。
aHUS临床病理表现与其他病因导致的TMA类似,但其肾脏受累表现较为突出。aHUS的发病机制主要为遗传性或获得性因素导致补体旁路活化的调节异常,补体过度激活而引起内皮细胞损伤,继而形成微血栓。遗传因素多为补体旁路调节蛋白如 H因子、I因子或MCP基因突变,导致细胞表面对补体旁路的抑制作用减弱;也可为C3或B因子基因突变导致旁路C3转化酶活性增强或对调节因子产生抵抗。获得性因素可由于产生抑制性抗补体旁路调节蛋白如H因子的自身抗体所致。本例患者即为抗 H因子抗体相关的aHUS。另外,值得一提的是,抗H因子抗体相关aHUS更多见于儿童,本例成人患者实属少见,可能与其类风湿关节炎的自身免疫背景有关。
本例患者经过血浆置换和甲泼尼龙治疗后,血清C3上升,病情稳定;但肾功能未恢复,最终进入终末期肾脏病。aHUS的治疗目前首选血浆置换治疗;美国和欧洲已批准抗C5单抗(eculizumab)作为治疗aHUS的一线用药,该药多用于血浆置换无效、血浆置换依赖或复发的患者。另有病例报道,抗H因子抗体阳性的aHUS可使用激素和(或)免疫抑制剂、抗CD20单抗等治疗。
aHUS预后较差,3年内死亡和ESRD总发生率为53%,需积极早期发现及干预。
1.aHUS是由于遗传或获得性因素造成补体旁路调节异常、补体旁路过度激活所致。
2.aHUS治疗首选血浆置换,效果差或复发者可使用eculizumab。
3.抗H因子抗体引起的aHUS可应用免疫抑制疗法。
(于 峰 刘 刚)
1.Barbour T,Johnson S,Cohney S,et al.Thrombotic microangiopathy and associated renal disorders.Nephrol Dial Transplant,2012,27(7):2673-2685.
2.Dragon-Durey MA,Loirat C,Cloarec S,et al.Anti-Factor H autoantibodies associtated with atypical hemolytic uremic syndrome.J Am Soc Nephrol,2005,16(2):555-563.
3.Józsi M,Strobel S,Dahse HM,et al.Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome.Blood,2007,110(5):1516-1518.
4.Noris M,Caprioli J,Bresin E,et al.Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype.Clin J Am Soc Nephrol,2010,5(10):1844-1859.
5.Song D,Liu XR,Xiao HJ,et al.The Clinical and Laboratory Features of Chinese Han Anti-factor H Autoantibody-associated Hemolytic Uremic Syndrome.Pediatric Nephrology,2017,32(5):811-822.









