


 收藏
收藏 已收藏
已收藏女性,27岁。
肉眼血尿3个月。
3个月前患者无明显诱因出现全程肉眼血尿,无尿路刺激症状及发热。10天前我院门诊查,尿检:蛋白 1.05g/d,RBC 满视野/HP,70%变形,WBC 1~3/HP;血生化:ALB 42.6g/L,SCr 70μmol/L;1天前开始口服氯沙坦(科素亚)50mg,每日1次,为进一步诊治收入院。发病来,患者无皮疹、关节肿痛、光过敏或口腔溃疡。
无特殊。
血压100/70mmHg,HR 75次/分,一般情况好,心肺腹查体未见明显异常;双下肢未见水肿。
尿检验:蛋白(+),定量0.53g/d;RBC 满视野/HP(大小不等,变形),WBC 0~2/HP;血清ALB 40.6g/L,SCr 72μmol/L;eGFR 110.4ml/(min·1.73m2)。血常规:WBC 5.92×109/L,Hb 129g/L,PLT 266×109/L。 ESR 9mm/h;CRP<1.00mg/L;RF<20.0U/ml;ASO 41.90IU/ml(正常<200);免疫球蛋白和C3、C4正常;ANA、抗ds-DNA抗体及抗ENA谱均阴性;乙肝五项均阴性。抗HCV抗体和抗HIV抗体均阴性;ANCA和抗GBM抗体阴性。
双肾 B 超:右肾11.9cm×4.4cm×3.3cm,实质厚 1.5cm;左肾 11.8cm×4.5cm×3.4cm,实质厚1.6cm;双肾实质回声偏强,内部结构不清晰。
慢性肾炎综合征 慢性肾脏病1期
患者青年女性,临床主要表现为肉眼血尿、少量蛋白尿(定量曾超过1g/d),肾功能和血压正常,经血管紧张素受体拮抗剂(ARB)治疗后尿蛋白减少;临床符合慢性肾炎综合征。结合患者临床表现及辅助检查,目前无继发性肾小球疾病证据,考虑为原发性肾小球疾病,下一步需肾活检明确病理类型,并指导进一步治疗和判断患者预后。
入院第4天行肾活检。免疫荧光:6个肾小球,C3(+++),余阴性,系膜区、节段性毛细血管壁颗粒样沉积(图1)。
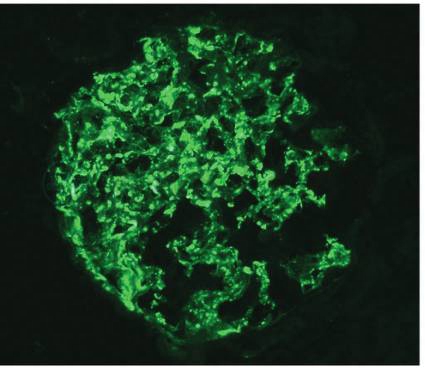
图1 直接免疫荧光可见C3在系膜区、节段毛细血管壁颗粒样沉积(×200)
光镜下可见46个肾小球,肾小球系膜细胞和基质轻度弥漫增生,局灶节段性中度加重伴内皮细胞增生,系膜区嗜复红蛋白沉积,其中3个细胞性、6个小细胞纤维性新月体形成,2个节段性硬化;肾小管上皮颗粒变性,灶状萎缩伴红细胞管型;肾间质灶状淋巴和单核细胞浸润伴纤维化;小动脉管壁增厚;符合局灶增生性肾小球肾炎(图2)。
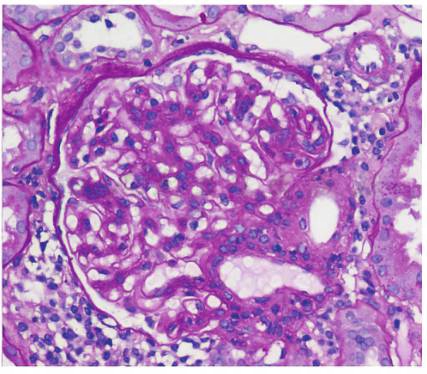
图2 光镜可见系膜细胞增生伴节段内皮细胞增生(PAS,×200)
电镜下可见8个肾小球,肾小球系膜细胞和基质轻度增生,节段性内皮细胞增生,系膜区团块状电子致密物沉积,基底膜未见病变,上皮足突节段融合;肾小管上皮溶酶体增多;肾间质淋巴单核细胞浸润(图3)。结合免疫荧光和光镜检查,符合C3肾小球肾炎。
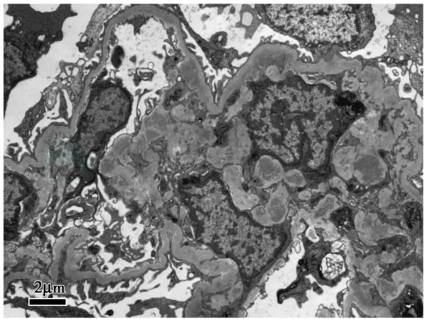
图3 电镜可见系膜区电子致密物沉积(×8 000)
鉴别诊断:①感染后毛细血管内增生性肾小球肾炎恢复期:患者起病前无前驱感染史,无血清C3动态变化,ASO不高,电镜下在上皮下未见驼峰样电子致密物,且新月体在毛细血内增生性肾小球肾炎中少见,恢复期更不常见,因此可基本除外感染后毛细血管内增生性肾小球肾炎。②患者肾小球病变不均一,电镜下系膜区团块状电子致密物沉积,应注意IgA肾病可能(可能因技术因素免疫荧光IgA未染上)。因此,专门加做免疫组化IgA染色证实为阴性,除外IgA肾病。
进一步检测患者血清H因子浓度正常,抗H因子抗体阴性;I因子浓度正常。
入院后继续口服氯沙坦(科素亚)。对患者已随访3年,目前尿蛋白阴性,尿红细胞3~5/HP,血肌酐和血压正常。
C3肾小球肾炎 慢性肾脏病1期
本病例提供了一个局灶增生型C3肾小球肾炎范例。最近几年国际上才提出C3肾小球病(C3 glomerulopathy)疾病概念,定义为肾小球免疫荧光C3沉积为主(C3c较其他免疫分子≥++),不论电子致密物的沉积部位。C3肾小球病包括致密物沉积病(dense deposit disease,DDD)和C3肾小球肾炎(C3 glomerulonephritis,C3GN)。DDD表现为特征性电镜下肾小球基底膜致密层见均质飘带状电子致密物沉积;除DDD外的C3肾小球病基本上都被归为C3GN。C3GN的电子致密物可沉积在系膜区和(或)内皮下,少数还伴上皮下、肾小球基底膜内非连续性(与DDD肾小球基底膜内飘带样电子致密物有根本区别)电子致密物沉积;C3GN光镜表现差异大,可表现为系膜增生性肾小球肾炎、轻微病变、膜增生性肾小球肾炎、毛细血管内增生性肾小球肾炎、新月体性肾小球肾炎或硬化肾小球病。C3GN临床表现差异大,可出现不同程度的蛋白尿、血尿和肾功能受损、高血压。
在部分C3肾小球肾炎患者中发现补体旁路调节蛋白如H因子、I因子、膜辅助因子(membrane cofactor protein,MCP)、CFHR5或C3基因突变,部分患者血清中还发现了C3Nef或抗H因子抗体阳性,支持C3GN发病与补体旁路途径调节异常有关。对该患者已进行部分补体旁路调节蛋白检测,但未发现异常,是否存在其他补体旁路调节缺陷仍待进一步研究。
C3肾小球肾炎为少见病,且近几年才统一定义疾病,对于其治疗目前无相关临床试验。总体上来讲,其治疗包括一般性治疗和特殊治疗。一般性治疗包括控制血压、RAS阻滞剂减少尿蛋白和控制脂代谢紊乱。糖皮质激素和免疫抑制剂的疗效尚不确定,而且患者就存在补体天然免疫缺陷,加用上述药物还需考虑到增加感染的风险。需要今后更多地研究探讨。
针对补体旁路调节异常发病机制的治疗可能是将来的发展方向,包括①血浆置换:可以补充缺乏的补体调节蛋白(如H因子),去除相应抗体,有个别病例报道;②抑制补体活化:C5单抗(eculizumab)可抑制C5分解为C5a和C5b,从而阻断了C5a和膜攻击复合物的作用,目前有病例报道在部分C3肾小球肾炎中有效,但部分患者仍出现肾功能进展;动物实验也表明阻断C5a可以改善病情,但不能阻止病理改变;C3活化在C3肾小球肾炎中发挥更重要的作用,期待将来针对抑制C3活化的治疗方法;③免疫抑制剂和CD20单抗:用于自身抗体(如C3Nef、H因子自身抗体)阳性患者,目前尚无相关报道。
目前对于C3肾小球肾炎预后缺乏长期大规模观察,总体上来讲,短期预后较好;Ginesta等延长随访至7年以上,部分患者可发展至依赖透析,提示长期预后可能较差;但Orfila等对部分患者随访至10~17年时只有2/13例出现中度肾功能减退;上述差异可能与诊断标准、病情轻重不一有关。
1.C3肾小球病包括DDD和C3肾小球肾炎。
2.C3肾小球肾炎临床和病理表现多样。
3.其治疗与预后尚无肯定结论。
(喻小娟 刘 刚)
1.Fakhouri F,Frémeaux-Bacchi V,Noël LH,et al.C3 glomerulopathy:a new classification.Nat Rev Nephrol,2010,16 (8):494-499.
2.Sethi S,Nester CM,Smith RJ.Membranoproliferative glomerulonephritis and C3 glomerulopathy:resolving the confusion.Kidney Int,2012,81(5):434-4341.
3.Servais A,Frémeaux-Bacchi V,Lequintree M,et al.Primary glomerulonephritis with isolated C3 deposits:a new entity which shares common genetic risk factors with heamolytic uraemic syndrome.J Med Genet,2007,44(3):193-199.
4.Sethi S,Fervenza FC,Zhang YZ,et al.C3 glomerulonephritis:clinicopathological findings,complement abnormalities,glomerular proteomic profile,treatment,and follow-up.Kidney Int,2012,82(4):465-473.
5.Servais A,Noël LH,Roumenina LT,et al.Acquired and genetic complement abnormalities play acritical role in dense deposit disease and other C3 glomerulopathies.Kidney Int,2012,82(4):454-464.









