


 收藏
收藏 已收藏
已收藏男性,43岁。
双眼睑、双下肢水肿40余天,发现血肌酐升高20天。
40余天前无诱因出现双眼睑、双下肢水肿,伴尿色加深,尿量正常。当地医院查血压正常,尿常规PRO(+++),红细胞80~100/HP,予“青霉素”治疗 5天后,双下肢水肿减轻。查尿蛋白定量 10.47g/d,SCr 107.9μmol/L,ALB 25.8g/L,IgG 3.56g/L,C 31.87g/L,RF、ANA及ANCA均阴性。1个月前于外院行肾穿刺,病理示免疫荧光IgG(++),C3(+),沿毛细血管壁线样沉积。光镜示多数新月体形成。电镜符合Ⅰ期膜性肾病。予甲泼尼龙冲击治疗3次(具体不详),环磷酰胺1g静脉输注,继之以泼尼松60mg每日一次口服至今。患者眼睑及双下肢水肿逐渐加重,伴尿量减少,700~800ml/d。近20天SCr进行性升高至271μmol/L,为进一步诊疗收入院。
体健。吸烟25年,40支/日。偶有饮酒。
体温36.5℃,脉搏60次/分,血压140/90mmHg,呼吸22次/分。听诊双肺呼吸音粗,未及干湿啰音。心脏和腹部查体未见明显异常。双下肢重度水肿。
借阅外院肾活检病理:免疫荧光 IgG(++),IgA(-),IgM(±),C3(+),FRA(-),C1q(-),沿毛细血管壁粗线样沉积(图1)。
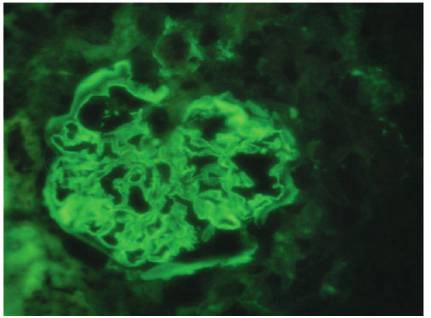
图1 膜性肾病合并抗GBM病:IgG沿肾小球毛细血管壁粗线状沉积(免疫荧光×400)
光镜可见23个肾小球,毛细血管袢严重破坏,伴多数新月体形成,9个细胞性、1个细胞纤维性新月体,1个节段性坏死。肾小管上皮颗粒及空泡变性,多灶状萎缩,肾间质多灶状淋巴单核浸润伴纤维化,小动脉管壁增厚(图2)。
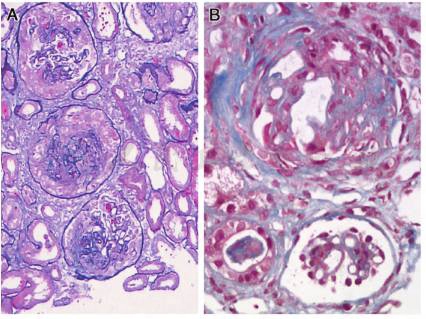
图2 膜性肾病合并抗GBM病:A:肾小球毛细血管袢破坏,新月体形成(PASM×200);B:肾小球上皮下嗜复红蛋白沉积,新月体形成(Masson×400)
结合荧光考虑新月体性肾炎Ⅰ型可能性大。电镜:肾小球基底膜轻度增厚,上皮下电子致密物沉积,上皮细胞足突融合,肾小管上皮细胞溶酶体增多,灶状萎缩,肾间质淋巴单核细胞浸润(图3),符合Ⅰ期膜性肾病。
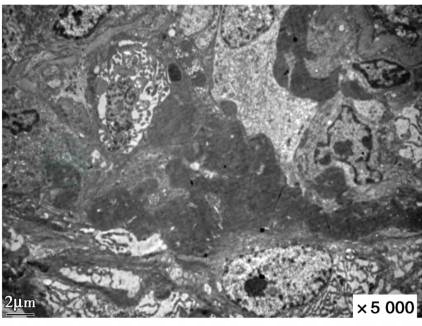
图3 膜性肾病合并抗GBM病:肾小球基底膜皱缩,上皮下电子致密物沉积,上皮细胞足突弥漫融合(电镜×8 000)
抗肾小球基底膜病急进性肾炎Ⅰ型急性肾损伤
肾病综合征膜性肾病
患者为中年男性,临床表现为肾病综合征,肾脏病理提示肾小球基底膜增厚,上皮下电子致密物沉积,上皮细胞足突融合,因此Ⅰ期膜性肾病诊断明确。同时患者临床表现有血尿,血肌酐进行性升高伴尿量减少,符合急进性肾炎综合征,结合肾脏病理免疫荧光可见IgG(++),C3(+),沿毛细血管壁线样沉积,以及光镜可见多数新月体形成,考虑抗肾小球基底膜病诊断明确。
尽管患者入院前已行甲泼尼龙冲击及静脉环磷酰胺等免疫抑制治疗,但血肌酐水平仍进行性升高,肾功能恶化。入院后查血清抗GBM抗体,结果为阳性,给予血浆置换5次,直至抗体转阴。同时继续激素治疗,将环磷酰胺改为50mg每日2次口服。患者尿量逐渐恢复正常,血肌酐降至139μmol/L。出院后规律随访,肾功能逐渐恢复正常,尿蛋白转阴。
抗肾小球基底膜病 急进性肾炎Ⅰ型 急性肾衰竭→CKD-2
肾病综合征 膜性肾病 慢性肾脏病2期
这是一个抗肾小球基底膜(GBM)病合并膜性肾病(MN)的病例。抗GBM病以原发性较为多见,但继发性抗GBM病也常有报道,其中以合并或继发于MN最为常见,其他也可见于IgA肾病、FSGS、膜增殖性肾炎等。自1974年以来,已有一系列关于抗GBM病合并MN的报道。这些病例大多是抗GBM病与MN同时发现,或是抗GBM病发生于MN之后,仅有个例患者抗GBM病发生于MN之前。
合并MN的抗GBM病患者,既有典型的抗GBM病特点,如循环和(或)肾组织中能够检出抗GBM抗体,肾脏可见多数新月体形成等,同时也有其特殊性。来自北京大学第一医院总结的资料发现,此类患者与典型的抗GBM病患者相比,尿蛋白水平较高,出现肾病综合征的比例较高;但是肾脏损伤的程度较轻,确诊时的血肌酐水平较低;出现少尿/无尿和肉眼血尿的概率较低;而且经过同样的血浆置换和强化免疫抑制治疗后,此类患者肾脏预后显著优于典型的抗GBM病的患者,1年的肾脏存活率可以达到62.5%,而后者仅有13.3%。因此,对于合并MN的抗GBM病患者,应该尽早明确诊断,及时给予和典型的抗GBM病相同的血浆置换和强化免疫抑制治疗,以期改善患者的预后。
合并MN的抗GBM病的独特临床表型可能与该类患者的自身抗体的免疫特性有关。来自北京大学第一医院的研究发现,此类患者抗GBM抗体所识别的抗原谱较窄,抗体水平较低,且抗体亚型以IgG4为主。关于两种疾病并存的原因有多种解释。较为普遍的理论是,患者罹患MN时,上皮下的免疫复合物沉积导致了GBM损伤,使GBM中原本处于遮蔽状态的自身抗原释放,从而导致了自身抗体的产生。已有研究证实MN中上皮下的钉突的重要成分就是Ⅳ型胶原的α3链和α4链,它们正是抗GBM抗体所识别的主要靶抗原。新近有动物实验发现,将少量α3(Ⅳ)NC1免疫DBA/1小鼠后,循环中出现抗GBM抗体,但肾脏病理为MN,如果增加免疫次数和剂量,则实验鼠可出现新月体性肾炎,提示抗GBM病和MN可以是对相同抗原产生的不同免疫反应,进而导致不同的疾病表型。
1.合并或继发于MN的抗GBM病是常见的继发性抗GBM病。
2.合并MN的抗GBM病患者临床多表现为肾病综合征,肾脏损害程度较轻,肾脏预后较好。
3.该类患者应给予和典型的抗GBM病相同的血浆置换和强化免疫抑制治疗,以期改善预后。
(崔 昭 陈 旻)
1.Basford AW,Lewis J,Dwyer JP,et al.Membranous nephropathy with crescents.J Am SocNephrol,2011,22(10):1804-1808.
2.Zhang JJ,Malekpour M,Luo WT,et al.Murine membranous nephropathy:immunization with alpha3(IV) collagen fragment induces subepithelial immune complexes and FcγR-independent Nephrotic Syndrome.J Immunol,2012,188(7):3268-3277.
3.Kim Y,Butkowski R,Burke B,et al.Differential expression of basement membrane collagen in membranous nephropathy.Am J Pathol,1991,139(6):1381-1388.
4.Minto AW,Kalluri R,Togawa M,et al.Augmented expression of glomerular basement membrane specific type IV collagen isoforms (alpha3-alpha5) in experimental membranous nephropathy.Proc Assoc Am Physicians,1998,110(3):207-217.
5.Jia XY,Hu SY,Chen JL,et al.The clinical and immunological features of patients with combined anti-glomerular basement membrane disease and membranous nephropathy.Kidney Int,2014,85(4):945-952.
6.Hopfer H,Hünemörder S,Treder J,et al.Glomerulopathy induced by immunization with a peptide derived from the goodpasture antigen α3Ⅳ-NC1.J Immunol,2015,194(8):3646-3655.









