


 收藏
收藏 已收藏
已收藏患儿,女,11岁。
主诉:咯血1天。
现病史:入院前1天于剧烈活动后突然出现阵咳,咳白色痰,痰中带血,约2ml,1小时后再次咳出约7~8ml鲜血,晨起时呕吐1次,为大量胃内容物,含少许血丝,以“咯血原因待查”急诊入院。病来低热,体温37.5℃左右,偶咳,无喘息,无胸痛胸闷及呼吸困难。近1年常有“感冒”咳嗽,不爱活动、乏力,但无低热、盗汗,无明显消瘦;无腹痛、腹泻,无鼻出血史。不偏食。无特殊药物服用史。精神欠佳,无黑便,无血尿、少尿。
既往史:近1年发现面色白有贫血,于当地诊断为“营养性贫血”。无心脏病、异物吸入等其他疾病史。无湿疹史。
过敏及接触史:无食物及药物过敏史。无肝炎、结核等传染病接触史。
个人及家族史:生长发育同同龄儿,按时进行预防接种,无月经初潮。独生子女,父母及一、二级亲属无类似咯血及出血性疾病史,肝炎、结核等家族史。
入院查体及相关检查:神志清楚,精神萎靡,面色略苍白,呼吸略促,约36次/min,口周无发绀;结膜略苍白,口唇白,咽红,咽后壁无血迹,扁桃体Ⅰ度;双肺叩诊清音,听诊呼吸音粗,右肺可闻及密集中小水泡音;心、腹及神经系统查体未见明显异常;四肢活动自如,甲床略苍白,无杵状指/趾,肢端温暖,双下肢无水肿,CRT<3秒。
辅助检查:门诊急检血常规:WBC 9.0×109/L,NE%87.4%,RBC 3.5×1012/L,HGB 66g/L,HCT 22%,MCV 64fl,MCH 20.9pg,MCHC 309g/L,PLT 330×109/L。门诊肺CT:右肺门及中下肺野弥漫性高密度及磨玻璃密度斑片影,左肺散在模糊斑片影。
入院后因诊断尚不明确,根据病情分析完善相关检查,同时给予对症支持治疗:①卧床休息,低流量吸氧,监护;②入院后发热伴有咳嗽,头孢呋辛和阿奇霉素控制感染,给予祛痰止咳药物。
检查及结果分析:
1.入院后完善各项检查 血常规如前所示,提示中度小细胞低色素性贫血,网织红细胞增多;尿常规正常;便常规正常,潜血阴性;肝肾功能正常,血清胆红素正常。血气分析正常。3次痰涂片未查到HLMs。
2.血CRP 12.1mg/L(0~8mg/L);ASO<25.0(0~200U/ml);ESR 19mm/h,均正常。病原学检测除肺炎支原体抗体(MPAb)1:320外,肺炎支原体抗体-IgM(MPAbIgM)阴性、咽拭子肺炎支原体DNA测定阴性,肺炎衣原体抗体-IgM、结核抗体(TBAb)阴性,常见呼吸道、肠道病毒及肝炎病毒检测均阴性,血细菌培养未见细菌生长,结核菌素试验阴性,提示除既往有肺炎支原体感染外,无结核等其他感染迹象。
3.贫血系列 促红细胞生成素(EPO)>824.00mU/ml(2.59~8.5mU/ml)增高,铁蛋白71.8ng/ml(11~336.2ng/ml),维生素B12 141pg/ml(180~914pg/ml),叶酸3.84ng/ml(3~17ng/ml)均正常;血清铁3.5µmol/L(7~30µmol/L)降低,提示血清铁和血清铁蛋白浓度降低,符合IPH的铁代谢改变。直接Coombs试验阴性,血清胆红素正常,除外免疫性溶血性贫血。骨髓穿刺:增生明显活跃骨髓象,粒红比例减低,未找到LE细胞。
4.免疫学检测 免疫球蛋白:IGG 12.3g/L(正常6.95~15.15g/L),IGA 1.76g/L(正常0.97~3.2g/L),IGM1.84g/L(正常0.4~1.59g/L)。淋巴细胞亚群:总T细胞(%)58(55~84),T抑制毒细胞(%)25(13~41),T 辅助细胞(%)29(31~60),Th/Ts 1.16(0.71~2.78),NK细胞(%)3(7~36),总 B 细胞(%)38(5~20)均正常,除外免疫缺陷病。总IgE 42.47U/ml正常。
5.RF、抗心磷脂抗体(ACA)、抗中性粒细胞胞质抗体测定(ANCA)及抗核抗体系列(ANA)均阴性,初步除外结缔组织病引起肺泡出血的可能。
6.心电图正常,心脏、肝胆脾及肾脏彩超未发现异常。
7.纤维支气管镜检查 右肺各支开口位置正常,黏膜粗糙、无明显分泌物附着,右中局部灌洗未见明显分泌物吸出,管壁毛糙,各支通气可,灌洗液呈洗肉水样;左肺各支开口位置正常,黏膜粗糙,无明显分泌物附着。BALF检查:一般细菌、结核菌及真菌涂片检查均未找到相应阳性菌,未查到肺含铁血黄素巨噬细胞。细胞学检查:分叶核细胞54%,淋巴细胞11%;巨噬细胞35%;细菌培养未见细菌生长。
8.肺部HRCT 入院后做肺部HRCT(图1),表现为双肺弥漫性高密度渗出性病变,以右肺显著。
以上辅助检查结果高度疑似“肺含铁血黄素沉着症”,但未查到“肺含铁血黄素巨噬细胞”。患儿于入院后4天热退,未再出现咯血,家长拒绝进一步做胃液含铁血黄素巨噬细胞等检查,不同意激素治疗,出院。
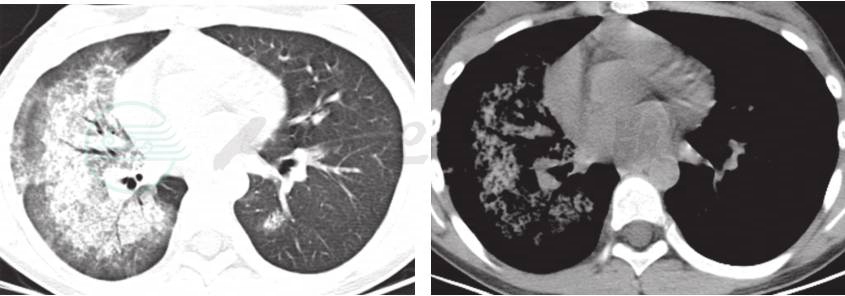
图1 入院后入院后完善肺部HRCT,提示:双肺弥漫性高密度渗出性病变,以右肺显著
1年后患儿再次因“低热,咳嗽伴气促3天,咯血1天”为主诉入院,期间反复有低热、咳嗽、气促表现,给予中药治疗,复查肺CT 4次,肺部浸润影时好时坏(图略)。入院查体仍有明显贫血貌,血常规:WBC 6.4×109/L,NE%60.0;RBC 2.9×1012/L,HGB 53g/L,HCT 18.1%,MCV 63fl,MCH 18.5pg,MCHC 294g/L,PLT 345×109/L,RC 3.74%。肺 HRCT(图2)显示双肺多发性弥漫、多发渗出性病变。入院后再次行纤维支气管镜检查,BALF中查到含铁血黄素巨噬细胞(图3),同时胃液中亦查到含铁血黄素巨噬细胞(图4)。确诊为:特发性肺含铁血黄素沉着症。给予对症支持治疗,输悬浮红细胞1次纠正贫血并予糖皮质激素治疗,口服甲泼尼龙片16mg,每天3次[1.6mg/(kg·d)],5天后无咯血症状,肺部CT(图5)浸润影明显吸收,出院。出院医嘱:①继续口服甲泼尼龙片16mg,每天3次,共3周;②碳酸钙D3片600mg,每天1次口服;③3周后小儿呼吸科门诊复诊。
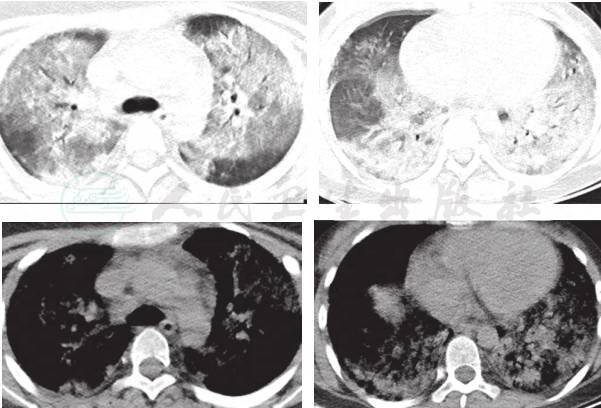
图2 1年后肺CT;双肺多发性弥漫、多发渗出性病变
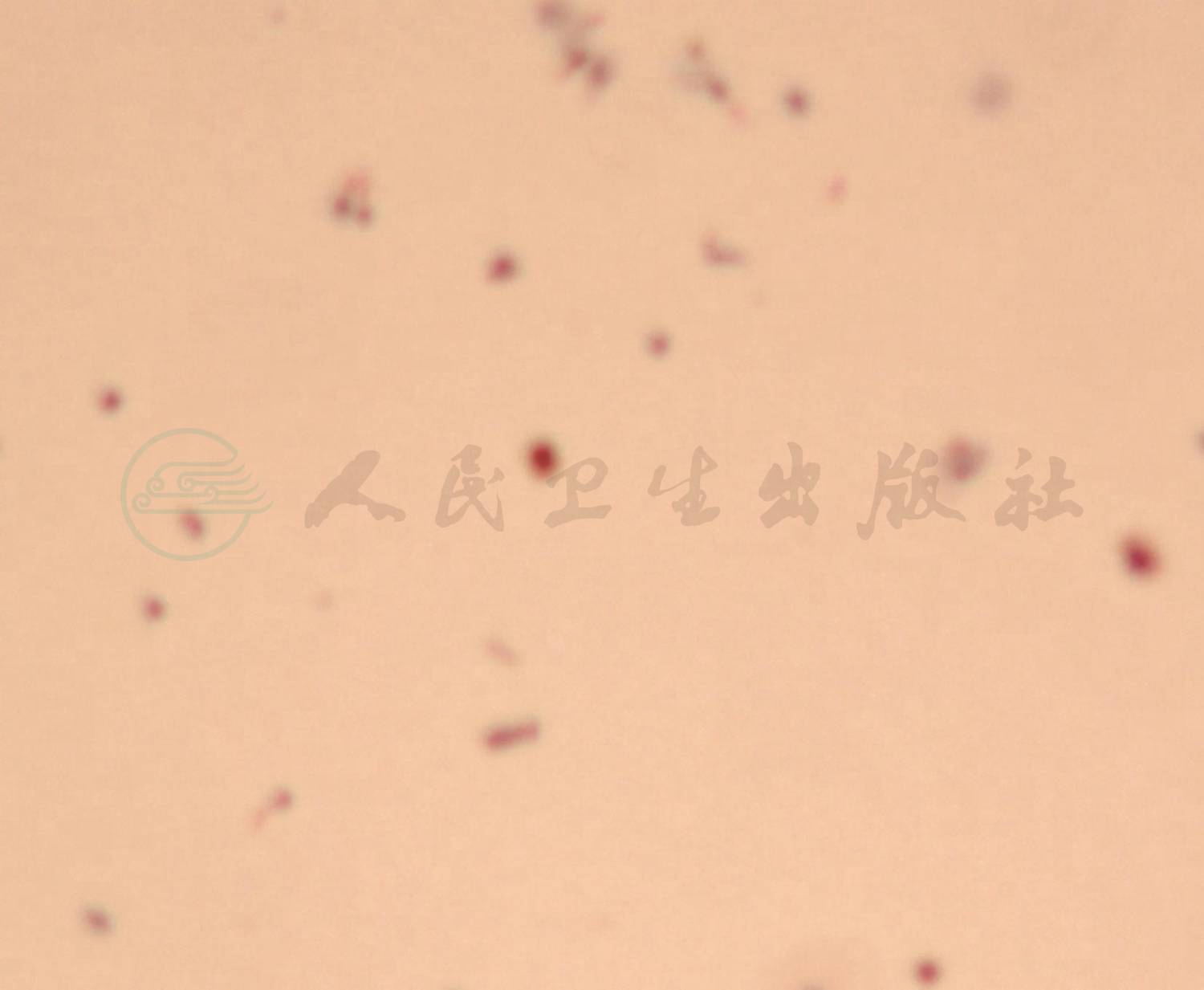
图3 肺泡灌洗液中查到含铁血黄素巨噬细胞

图4 胃液中查到含铁血黄素巨噬细胞
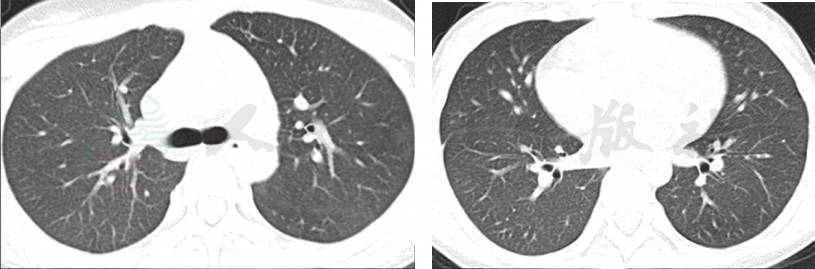

图5 治疗后复查肺CT
出院1个月后再次因“间断发热2天,咳嗽咯血1天,呼吸困难15小时”入院。上次出院后按医嘱口服甲泼尼龙片(每次4片,每天3次),无咯血症状和不适,3周后未复诊,并自行停药。入院查体:体温37.8℃;脉搏152次/min;呼吸70次/min;血压105/75mmHg;体重43kg。神志清楚,状态差,精神萎靡,满月脸,面色苍白,呼吸急促,口唇发绀,伴有明显鼻翼扇动和三凹征,双肺散在哮鸣音,双肺底密集细小水泡音,心率快,余未见异常。入院后处理:①急检血常规、CRP、DIC常规、血气分析,急查肺CT等,肺CT示双肺弥漫性渗出性病变;②心电、血氧、血压监护,面罩吸氧;③甲泼尼龙 40mg[1.6mg/(kg·d)],12小时1次,静脉点滴5天,改40mg每天1次,静脉点滴9天,后改口服甲泼尼龙片16mg,每天2次;④血常规:WBC 7.6×109/L,NE%83.5%,CRP 133mg/L,予头孢噻肟那舒巴坦钠控制感染;⑤酚磺乙胺、维生素K1止血。入院第5天患儿状态明显好转,呼吸平稳,无鼻翼扇动及三凹征,无发绀,双肺水泡音消失,肺CT示(图6)双肺弥漫病变部分吸收,肺部透过度略改善,2周后出院。出院医嘱:①口服甲泼尼龙片16mg,每天2次;②小剂量红霉素100mg/次,每天2次[5mg/(kg·d)];③乙酰半胱氨酸600mg/次,每天 1次口服;④碳酸钙D3 600mg/次,每天2次口服;⑤槐杞黄颗粒10g/次,每天2次,口服3个月;⑥2周后门诊复查。
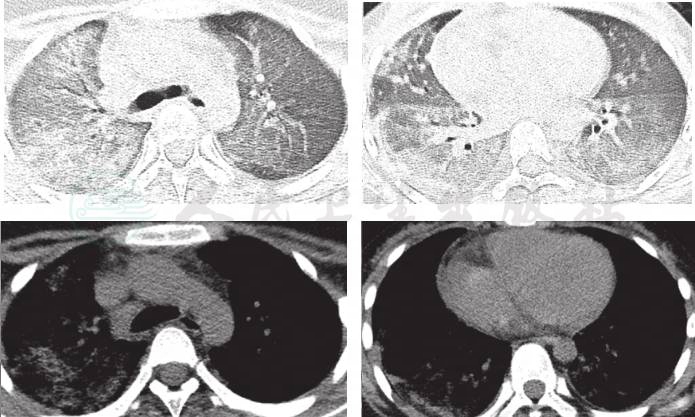
图6 返院后复查肺CT
2周后门诊复查,无咳嗽咯血,查体:血压110/75mmHg,库欣面容,呼吸平稳,双肺呼吸音清无啰音。血常规RBC 3.5×1012/L,HGB 110g/L;肺CT双肺野透过度下降,散在分布斑片状磨玻璃密度影,双肺病变明显好转(图略)。甲泼尼龙片每天总量8片(32mg)隔天晨顿服,每2周减半片,余用药同前,每月复查1次。6个月后甲泼尼龙片减至2片隔天晨顿服,加用布地奈德0.5mg,每天2次雾化吸入治疗,停用小剂量红霉素,余治疗不变,维持治疗1年,期间曾患化脓性扁桃体炎、肺炎各住院1次,无咯血现象,感染期间除控制感染外给予甲泼尼龙40mg,每天2次静脉点滴3天,其后继续口服甲泼尼龙片2片隔天晨顿服维持治疗,监测血常规、血压、血糖、骨密度及肺功能,观察治疗中。
特发性肺含铁血黄素沉着症。
诊断依据:①有反复发作性咳嗽、咯血和贫血症状、体征,血常规提示中度小细胞低色素性贫血;②发作时肺部闻及水泡音,肺HRCT以双肺弥漫性肺浸润和肺间质的改变;③BALF中查到含铁血黄素巨噬细胞,同时胃液中亦查到含铁血黄素巨噬细胞;④除外其他肺出血性疾病,如血管炎、风湿性疾病、免疫缺陷病、肺结核、支气管异物、血管畸形和反复支气管肺炎等。
IPH最先于1864年由Virchow报道,是一种少见的无肺泡毛细血管炎的弥漫性肺泡内出血疾病(diffuse alveolar hemorrhage disease,DAH),以肺间质含铁血黄素沉着为显著特点,好发于10岁以下儿童,以6岁以下居多。国外文献报道的年发病率为0.24/10万~1.23/10万,占儿童肺间质性肺疾病的8%,儿童期男女发病率无明显差异,成人中男性居多。目前我国尚无确切的发病率统计。典型的IPH临床表现为咯血、贫血、弥漫性肺浸润“三联症”,痰、胃液或支气管肺泡灌洗液检查可见含铁血黄素细胞。本病的诊断为排他性诊断,因此有条件地尽量争取肺活检,以除外其他疾病,尤其是肺泡毛细血管炎所致的弥漫性肺泡出血症。在中国儿童间质性肺疾病中,IPH可能是首位的病因。IPH引起的急性肺出血是临床上的危重症,病情凶险,救治难度大,但随着对IPH治疗经验的积累,其预后已较既往有所改善。
IPH病因未明,可能与免疫、遗传、牛乳过敏、环境等因素相关,但缺乏进一步的确切依据。已有一些家族性病例的报道,提示该病可能存在遗传背景,但未发现与本病相关的候选致病基因。发病机制尚未明确,由于IPH患儿对肾上腺皮质激素和/或免疫抑制剂治疗显示出良好的近期效果,由此提示免疫机制参与了疾病的发生发展过程。多数学者认为,抗原-抗体复合物介导的肺泡自身免疫性损伤,致使肺泡毛细血管通透性增加,导致肺小管出血可能是IPH最为重要的发病机制。文献报道IPH患儿中部分出现外周血T细胞亚群异常及免疫球蛋白增高。有学者观察到,临床上约有25%生存期超过10年的IPH患者相继发生如自身免疫性甲状腺炎、自身免疫性溶血性贫血、幼年特发性关节炎等自身免疫性疾病。另有IPH患儿同时合并麦胶性肠病(又称乳糜泻),血清中麦胶蛋白的IgG、IgA抗体测定滴度增高。但有关IPH发病机制的研究很少。
IPH主要病理改变为肺泡毛细血管出血,当肺泡毛细血管反复出血,渗出的血液溶血后,其中珠蛋白部分被吸收,血红蛋白转化为含铁血黄素沉着于肺组织。含铁血黄素被巨噬细胞摄取,这些巨噬细胞产生早期炎性介质;如果反复出血,将导致慢性炎症及纤维化。咯血或呕血是血流入肺泡腔的间接表现,失血及肺组织中铁的沉积导致缺铁性贫血。晚期可出现严重的肺纤维化、肺心病。
本病临床症状和病程取决于肺内出血的程度及期限,多数病程长,发作与自动缓解交替出现。依据临床病程将IPH分为3期:急性出血期、慢性反复发作期、静止期或后遗症期。
1.急性出血期
突然发病,咳嗽、咯血、气促,可有面色苍白、疲乏等表现,而咯血最具有诊断意义,咯血可呈痰中带血,也可大量咯血。小儿不会咳痰常无咯血,常以面色苍白为主要表现,有时伴呕血、黑便或轻度黄疸,久之出现疲乏、食欲缺乏、生长发育落后。严重病例可呈大咯血表现,导致急性呼吸衰竭,部分死于出血性休克或出血合并感染。合并感染时出现发热。此期肺部体征不尽相同,可无体征或少许湿啰音、哮鸣音,也可有肺部实变体征。贫血患儿可出现心尖部收缩期杂音。
胸部X线检查两肺野透亮度普遍减低,呈毛玻璃样改变及大片云絮状阴影,以肺门及中下肺野多见,两侧多对称分布,肺尖、肋膈角及肺底表现较轻甚至不累及。在HRCT上,急性肺出血往往表现为片状磨玻璃样阴影或实变。肺部病变经治疗后多在1~2周内明显吸收,有时可延续数月或反复出现。
2.慢性反复发作期
以长期或反复咳嗽、咯血、胸痛、哮喘及低热为特征,由于反复的肺出血,大量含铁血黄素在肺内沉积,并由咳痰丢失,慢性失血导致缺铁性贫血,明显的贫血貌、心悸、乏力,部分患儿出现肝脾大、杵状指/趾。
胸部X线检查两肺广泛分布的小结节影及细小的网状影。随着病变进展网状影渐渐增多变粗。若有新鲜出血,则在细网状影的基础上,同时有磨玻璃影出现。
3.静止期或后遗症期
稍有咳嗽、气促,常无咯血或贫血。病程后期因反复出血形成广泛间质纤维化,出现杵状指/趾、肺功能不全、肺动脉高压、肺源性心脏病和呼吸衰竭。
胸部X线检查肺纹理增多而粗糙,可有小囊样透亮区或纤维化改变,并可出现肺动脉高压和肺心病的X线征象。小叶间隔增厚和弥漫性小结节为亚急性期及慢性期表现。
本病的诊断要点如下:①原因不明的小细胞低色素性贫血;②反复咳嗽、气促,伴或不伴咯血;③胸片或CT可见急慢性肺浸润;④痰、胃液或BALF检查可见含铁血黄素细胞;⑤肺组织活检可见含铁血黄素沉积及不同程度的纤维化,但无肺泡毛细血管炎;⑥排除其他原因的肺出血。值得注意的是,在一些长期随访病例中,原诊断IPH的患者若干年后表现为肺肾出血综合征、系统性红斑狼疮等疾病,因此,IPH的诊断必须建立在除外继发性肺含铁血黄素沉着症的基础上,注意和结缔组织疾病、原发或继发性血管炎等疾病鉴别,必要时可行肺活检,以确定是否存在肺泡毛细血管炎所致的弥漫性肺泡出血。
糖皮质激素目前仍是治疗首选药物,急性期尽早使用激素能改善症状,降低毛细血管的通透性,迅速起到止血作用,从而保护肺功能和减少肺纤维化形成。但其剂量和疗程,尤其是疗程仍缺乏循证医学的证据,推荐剂量泼尼松1~2mg/(kg·d)口服治疗2个月,逐渐减量至最低控制症状维持量,持续治疗6个月或更长时间。对严重威胁生命的IPH患儿,甲泼尼龙 10~20mg/(kg·d),静脉滴注,连用3天,病情缓解后改为口服泼尼松1~2mg/(kg·d),逐步减量同上;症状较重者,X线病变未静止及减药过程中有反复的患儿,疗程应延长至1~2年,停用激素指征:临床及实验室检查恢复至少1年。另外,目前国内外均有采用激素吸入性疗法取得一定疗效的报道,当口服激素使病情缓解,为减轻全身应用激素带来的不良反应时,可在口服激素逐步减量到一定维持剂量并控制症状3个月左右加吸入激素治疗。对肾上腺皮质激素效果不佳或激素剂量依赖、肺功能持续下降的患儿可考虑选用免疫抑制剂治疗。
既往认为IPH预后较差,患者通常死于急性肺出血和呼吸衰竭。近年的病例报道显示5年生存率是86%。IPH预后的关键在于早期诊断,尽早控制急性发作,减少复发,保存肺功能,否则贻误时机,肺功能出现不可逆改变。尽早诊断、合理地运用药物治疗可能有助于改善预后。
(蔡栩栩)









