


 收藏
收藏 已收藏
已收藏患儿,男,4个月。
主诉:咳嗽10余天,发热5天,气促3天,皮下硬结2天。
现病史:入院前10余天出现咳嗽;入院前5天即接种卡介苗后第2天出现发热,体温39.2℃,接种部位皮肤未见异常;入院前3天明显气促,于当地医院静滴头孢呋辛钠3天、甲泼尼龙2天(剂量不详)无好转;入院前2天出现皮下多发硬结,以双下肢较多。
既往史:患儿生后24天时左耳后皮下脓肿,流黄色脓汁;1.5个月时患急性支气管肺炎。
过敏及接触史:无食物及药物过敏史。无肝炎、结核等传染病接触史。
个人及家族史:G1P1,足月剖宫产娩出,生后无窒息。出生体重3.45kg,母乳喂养。既往患儿生长发育同同龄儿。母亲的舅舅生后不久夭折。
入院查体及相关检查:体温38.6℃;脉搏180次/min;呼吸82次/min;血压(下肢)95/57mmHg;体重6.5kg。精神萎靡,反应差,呻吟,面色灰白。全身见6个大小不等的皮下硬结,双下肢为多,直径0.8~3.2cm不等,中心紫红色,部分有破溃结痂,周围绕以红晕。患儿周身浅表淋巴结未触及。前囟平软。双鼻腔阻塞,见黄色混浊液体流出。呼吸急促,鼻翼扇动及三凹征阳性,口周明显发绀,双肺闻及少许中小水泡音。肝肋下5cm,脾肋下1cm,质地均匀,Ⅰ度。指/趾端明显青紫。神经系统查体无阳性体征。
辅助检查:门诊急检血常规:WBC 6.1×109/L,NE%24.7,L%29.08,RBC 3.83×1012/L,HGB 119g/L,PLT 320×109/L。CRP 128mg/L。门诊胸片:支气管肺炎、胸腺缺如?
检查及结果分析:
1.入院后完善各项检查 血常规:WBC 5.8×109/L,N 55.7%,L 19.1%,RBC 3.60×1012/L,Hb 112g/L,PLT 134×109/L。尿常规正常。便常规正常,潜血阴性。肝肾功能正常,血清胆红素正常。血气分析:pH 7.412,PCO2 30.0mmHg,PO2 51.1mmHg;血乳酸2.3mmol/L;血氨3.0mmol/L。
2.血CRP 121mg/L(0~8mg/L);ASO<25.0(0~200U/ml);病原学检测肺炎支原体抗体、肺炎支原体抗体-IgM(MPAbIgM)阴性、咽拭子肺炎支原体DNA测定阴性,肺炎衣原体抗体-IgM、结核抗体(TBAb)、常见呼吸道、肠道病毒、TORCH病毒及肝炎病毒检测均阴性,结核菌素试验阴性,无结核等其他感染迹象。
3.血细菌培养、痰细菌培养及鼻腔分泌物细菌培养均培养出铜绿假单胞菌。
4.免疫学检测 免疫球蛋白:IGG 0.4g/L(正常6.95~15.15g/L),IGA<0.22g/L(正常0.97~3.2g/L),IGM< 0.18g/L(正常0.4~1.59g/L)。淋巴细胞亚群:总T细胞(%)29(55~84),T辅助细胞(%)4(31~60),Th/Ts 0.2(0.71~2.78),NK细胞(%)8(73~36),总B细胞(%)63(5~20),总IgE 32.1U/ml,正常。
5.心脏未发现异常;肝胆脾彩超:肝脾大(肝脏上界位于右锁中线第6肋间,肋下长约3.9cm,肝实质回声较均匀;脾肋间厚约1.7cm,肋下长约1.0cm)。
6.肺部CT(图1) 双肺段多发斑片影、结节影,以双肺下叶为著,未见胸腺影。
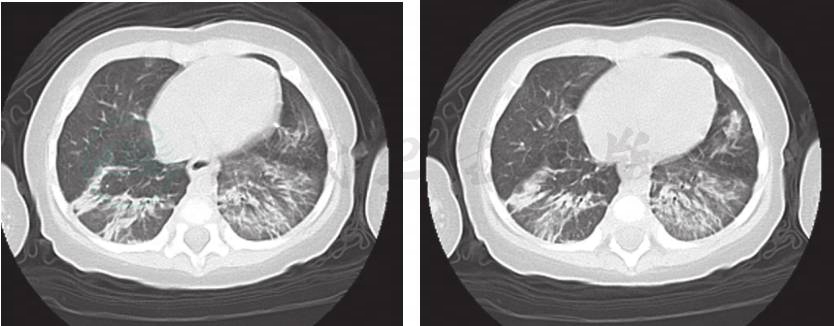
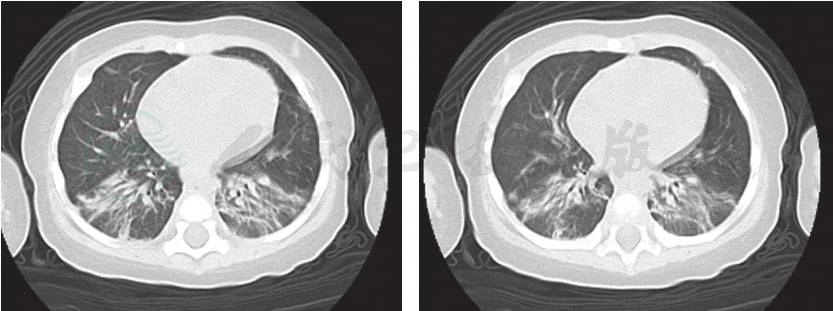
图1 入院后完善肺CT检查,提示:双肺段多发斑片影、结节影,以双肺下叶显著,未见胸腺影
患儿入院后给予对症治疗:亚胺培南西司他丁钠0.2g,每6小时1次,静滴10天;盐酸万古霉素65mg,6小时1次,静滴 10天;丙种球蛋白2.5g,每天1次,共8天。入院第4天患儿口腔咽后壁及软腭发现黄白脓性物,上软腭与鼻道间形成瘘,鼻腔有血性液体流出,而后转为黄白色脓性液体。鼻腔分泌物培养:铜绿假单胞菌;鼻腔分泌物涂片找到革兰氏阳性菌,腰穿脑脊检查未见异常。入院治疗2天体温平稳,于入院第6天体温再次升高为38.2℃。复查肺CT:双肺下叶渗出性病变较前吸收,双肺上叶出现结节状影,边缘有毛刺征。检测1,3-β-D葡聚糖1.33mmol/L,考虑合并真菌感染。予抗真菌治疗:伏立康唑40mg,每12小时1次,静脉滴注1天后改25mg,每12小时,共静脉滴注12天;第2天体温降37.2℃左右。住院治疗18天,复查血细菌培养阴性,复查肺CT:双肺炎症较前进一步吸收。于上海交通大学医学院附属新华医院行基因分析结果为T-B+SCID,受体γ链(γc)基因缺陷,X连锁的IL-2Rγ链缺陷。患儿母亲为携带者。故确诊为SCID。家长因经济原因最终放弃治疗。
1.先天性严重联合免疫缺陷病。
2.铜绿假单胞菌性败血症。
3.急性支气管肺炎(重症,细菌混合真菌感染)。
4.Ⅰ型呼吸衰竭。
5.鼻漏。
6.腮裂瘘。
7.多发皮下疖肿。
SCID是由遗传因素引起的淋巴干细胞发育成熟障碍,表现为外周血T淋巴细胞缺如或严重减少,并伴随其他淋巴细胞如B细胞功能异常。由于缺乏成熟的T细胞,机体丧失适应性细胞免疫功能,对多种病原体普遍易感,其中机会性致病原占主导地位。SCID分T细胞缺陷、B细胞计数正常或增高但功能存在异常(T-B+SCID)及T和B细胞均缺如(T-B-SCID)两种。T-B+ SCID遗传方式有X-连锁及常染色体隐性遗传两种,T-B-SCID均为常染色体隐性遗传。X-连锁SCID在美国最常见,占全部SCID的45%。其临床表现为口腔念珠菌病、红斑,持续性腹泻,多种条件致病菌感染及革兰氏阴性菌败血症等。SCID多在3个月内起病,生后6个月内多以慢性腹泻、间质性肺炎和/或顽固性皮肤黏膜念珠菌病为主,继而发生频繁的致死性严重感染。感染谱十分广泛,可为持续低毒力条件致病菌如卡氏肺孢子菌、CMV及隐孢子虫等感染。细菌感染以中耳炎、肺炎及皮肤感染多见,播散性卡介苗感染也较常见。真菌感染主要表现为鹅口疮、喂养困难和体重不增。SCID多伴生长发育障碍。此病预后差,多数患儿在1~2岁内死于各种严重感染并发症。
SCID胸部影像学重要特征是无胸腺影像,X线胸片或肺CT表现为胸腺缺如所致的上纵隔狭窄。SCID患儿几乎无淋巴细胞免疫功能,外周血淋巴细胞绝对计数往往少于1000/L,但淋巴细胞计数正常也不排除免疫缺陷。SCID患儿T淋巴细胞亚群改变明显,表现为T淋巴细胞数目减少或缺如,增殖反应低下。对考虑可能存在SCID应及时行免疫功能筛查,检测血常规、T细胞亚群、免疫球蛋白,如发现异常则应选择广谱杀菌性抗生素控制感染,并适当输注丙种球蛋白,若需输血,应输注25Gy辐照血,以防止移植物抗宿主反应。最佳的治疗手段是行造血干细胞移植术,以挽救患儿生命。
(王 佳 韩晓华)









