


 收藏
收藏 已收藏
已收藏患儿,男,8岁。
主诉:间断发热7天,咳嗽5天。
现病史:患儿7天前无明显诱因出现发热症状,最高体温40.0℃,每天发热1次,无寒战及抽搐,家属予患儿口服中药及头孢类药物(具体不详)2天,患儿热退。入院5天前患儿出现咳嗽,为声咳,有痰,不伴喘息。入院3天前患儿再次出现发热,起初体温波动于37.0~38.0℃,家长自予其口服美林后热可退,昨日体温升高至40.0℃,同时患儿咳嗽症状较前加重,故入院治疗。患儿病来精神状态良好,饮食、睡眠及大、小便正常。
既往史:患儿2岁时患“肺炎”1次,3岁开始每年上呼吸道感染3~4次、患中耳炎1~2次。
过敏及接触史:否认明确食物及药物过敏史。否认结核病及肝炎接触史。
个人及家族史:生长发育同同龄儿,按时进行预防接种。独生子女,父母体健;否认遗传代谢性病家族史。
入院查体及相关检查:神志清楚,状态好,周身皮肤未见皮疹及出血点,浅表淋巴结未触及肿大。呼吸平稳,右肺可闻及少许干鸣音。心音有力、律齐,各瓣膜听诊区未及杂音。腹软,肝脾肋下未触及,肠鸣音良好,四肢末梢温,肌张力正常,CRT<3秒,神经系统查体无阳性体征。
辅助检查:(笔者医院门诊)血常规:WBC 9.4×109/L,NE%74%,HGB 117g/L,PLT 187×109/L;CRP 21.3mg/L。胸片:右肺中叶炎症。
入院后予患儿完善相关检查,静脉滴注头孢孟多酯联合阿奇霉素抗感染,用药3天后患儿仍持续发热,病情未得到明显缓解。
入院后完善各项检查,结果及分析如下:
1.尿常规正常;便常规正常;肝、肾功能正常。
2.病原学检测 ASO<54.10(0~200U/ml);肺炎支原体抗体(MPAb)阴性,肺炎支原体抗体-IgM(MPAbIgM)阴性、咽拭子肺炎支原体DNA测定阴性,肺炎衣原体抗体-IgM、结核抗体(TBAb)、结核菌素试验、常见呼吸道、肠道病毒及肝炎病毒检测均阴性,血细菌培养未见细菌生长,结核菌素试验阴性,未提示有肺炎支原体、衣原体、结核及常见病毒等感染迹象。
3.免疫功能检测 免疫球蛋白:IgG<1.45g/L(正常6.95~15.15g/L),IgA<0.25g/L(正常0.97~3.2g/L),IgM 0.2g/L(正常0.4~1.59g/L)。淋巴细胞亚群:总T细胞(%)94.61(55~84),T抑制毒细胞(%)25.95(13~41),T辅助细胞(%)63.28(31~60),Th/Ts 2.44(0.71~2.78),NK细胞(%)5.17(7~36),总B细胞(%)0.2(5~20)。患儿免疫球蛋白IgG、IgA、IgM均明显降低,且总B细胞亦明显低于正常,考虑患儿存在体液免疫功能缺陷。
4.肺部CT检查(图1) 提示右肺中叶及下叶纹理增多、模糊,沿肺纹理走行可见多发腺泡结节、小斑片状模糊影,右肺中叶部分实变,内可见含气支气管。
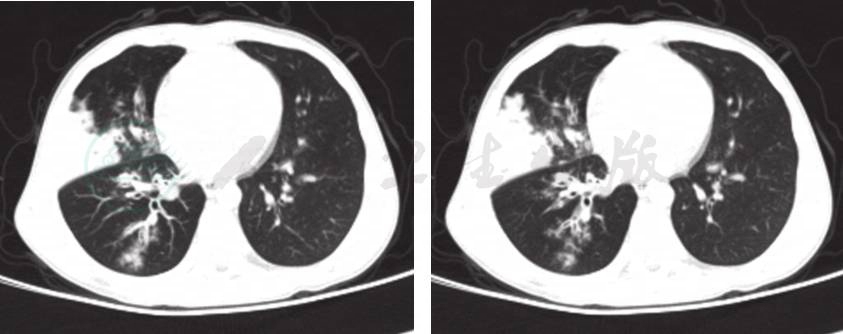
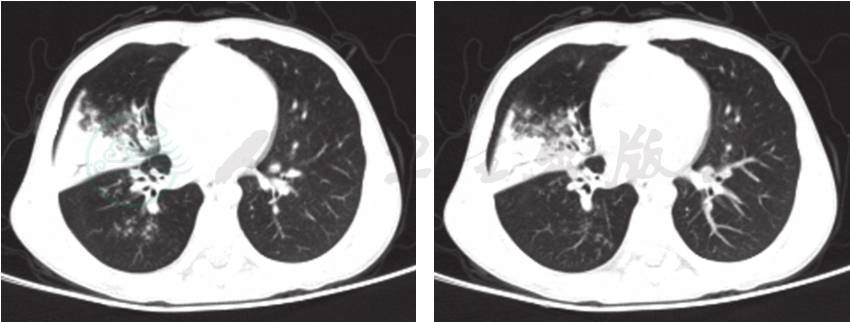
图1 入院后完善肺CT
提示:右肺中叶及下叶纹理增多、模糊,沿肺纹理走行可见多发腺泡结节、小斑片状模糊影,右肺中叶部分实变,内可见含气支气管
结合患儿化验检查,考虑患儿存在先天性体液免疫缺陷可能,进一步追问家族史,其母亲诉患儿有2个舅舅均在5~6岁时因病夭折,具体病因不详;故进一步考虑其为X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia,XLA)。立即予患儿静脉注射免疫球蛋白[400mg/(kg·d)]治疗,2天后热退,继续给予头孢类抗生素抗感染治疗1周后复查肺CT,炎症明显吸收后出院。
1.右肺中叶大叶性肺炎
诊断依据:①患儿急性起病,有发热及咳嗽的临床表现;②肺部查体右肺可闻及少许干鸣音;③肺部CT检查,提示右肺中叶及下叶纹理增多、模糊,沿肺纹理走行可见多发腺泡结节、小斑片状模糊影,右肺中叶部分实变,内可见含气支气管。
2.X连锁无丙种球蛋白血症
诊断依据:①患儿男孩。②既往患儿存在反复感染病史:患“肺炎”1次,3岁开始每年上呼吸道感染约3~4次,患中耳炎1~2次。③患儿母亲的2个哥哥均在5~6岁时因病夭折,具体病因不详。④免疫功能检测,免疫球蛋白:IgG<1.45g/L(正常6.95~15.15g/L),IgA<0.25g/L(正常0.97~3.2g/L),IgM 0.2g/L(正常0.4~1.59g/L);淋巴细胞亚群:总T细胞(%)94.61(55~84),T抑制毒细胞(%)25.95(13~41),T辅助细胞(%)63.28(31~60),Th/Ts 2.44(0.71~2.78),NK细胞(%)5.17(7~36),总B细胞(%)0.2(5~20);患儿免疫球蛋白IgG、IgA、IgM均明显降低,且总B细胞亦明显低于正常,考虑患儿存在体液免疫功能缺陷。故可临床诊断。
XLA需要与其他存在明显体液免疫缺陷的疾病相鉴别:
1.普通变异型免疫缺陷病(CVID) XLA和CVID在临床上都以反复感染为主且外周血IgG、IgA、IgM含量均普遍降低,但XLA的发病年龄通常较CVID早,临床症状通常较CVID患儿重,且XLA患儿的CD19(B细胞)均<2%,而CVID的CD19 大致正常,故CD19正常或降低是鉴别此两种疾病的关键点。
2.高IgM综合征(XHIM) 由CD40L基因突变引起,IgG、IgA降低,IgM增高或正常,据报道只有50%IgM增高。其与XLA都可表现为男性患儿且血清IgG水平低于相应年龄正常值至少2个标准差,但XHIM患儿的T细胞和B细胞数正常,故可鉴别。
目前全世界报道的XLA发病率约为(6~10)×10-6,美国为4×10-6。虽然我国从1999年已经开始原发性免疫缺陷病的登记工作,但目前尚无发病率的报道。XLA的病因为Bruton酪氨酸激酶(BTK)缺陷,导致外周血B淋巴细胞明显减少,使Ig合成不足,血清中各类免疫球蛋白明显降低或缺乏,对很多抗原不能产生特异性抗体反应,使机体的免疫力低下,容易发生细菌感染。但T淋巴细胞数量及功能正常。因此,外周血B细胞和血清免疫球蛋白明显下降是该病的主要实验室特征。1993年,XLA的致病基因被定位于BTK基因。目前,世界范围内已经报道超过600种BTK基因突变类型,包括错义突变、无义突变、插入、缺失和剪接位点突变等。因其为X连锁隐性遗传,完全传递,故仅为男性受累,且每例BTK突变患者均有症状。由于母体IgG可通过胎盘进入胎儿体内,故生后头2~3个月无症状,随着母体IgG的不断减少,患儿一般表现为生后4~12个月左右开始发生反复化脓感染,伴全身淋巴组织发育不良。
XLA患儿的感染部位以呼吸道为主,消化道亦是十分常见的受累部位。较多文献报道XLA患儿有关节受累表现,中国台湾省更是有报道称20%的XLA患者有关节炎表现。
目前认为只要具备反复细菌感染、血清免疫球蛋白明显降低及外周血CD19(B细胞)(或CD20)低于1%,即可诊断为原发性无丙种球蛋白血症,其中约80%为X连锁无丙种球蛋白血症,约15%~20%为非X连锁无丙种球蛋白血症。因目前临床上无法将两者区分,故应注意询问家族史。
XLA因为体液免疫缺陷,故患者主要以细菌感染为主,而且多迁延难愈,治疗感染应用大环内酯类等抑菌性抗生素疗效极差,以β-内酰胺类等杀菌为主的抗生素疗效较好。患儿常因体液免疫缺陷,自身抗细菌感染能力下降,常易发生化脓性细菌感染,如肺炎链球菌或嗜血流感杆菌,且往往不能及时应用有效抗生素治疗,而导致病情迁延不愈,并容易发生耐药菌感染。除及时和选择合适的抗生素抗感染外,目前主要治疗XLA的方法为静脉注射丙种球蛋白(IVIG)补充疗法。美国有报道称定期进行IVIG治疗,患儿的生活质量并不差。但因丙种球蛋白半衰期较短,需每月注射,其起始量一般为400~600mg/kg,然后需根据患儿对治疗的反应来调整用药剂量及给药间隔时间,即用药的剂量和频率必须个体化,使免疫球蛋白维持在正常的上限水平。当血清IgG谷浓度维持在5g/L以上时,可明显减少感染机会并能改善肺功能。虽然IVIG可明显降低患儿感染的频率和严重程度,却不能杜绝感染。对于病情严重的患者,可试行骨髓移植重建免疫治疗。
(王 佳 韩晓华)









