


 收藏
收藏 已收藏
已收藏患儿,男,9岁。
主诉:反复咳嗽伴黄绿痰1年余,加重6个月。
现病史:患儿入院前1年起出现咳嗽,咳嗽时伴有黄绿色黏痰,晨起严重。病程中未曾出现咯血。患儿入院前6个月,因咳嗽加重伴痰多,在当地医院住院治疗3周,胸部CT检查发现支气管扩张伴感染,经积极抗感染治疗后,咳嗽咳痰好转,予以出院。出院后仍有咳嗽伴咳黄绿痰,至本院进一步就诊,门诊以“支气管扩张”收治入院。平时无腹痛,无腹胀、腹泻,大便正常。病来患儿精神状态尚可,进食及睡眠正常,小便正常。
既往史:既往有反复鼻窦炎病史。既往无重症肺炎病史。
过敏及接触史:无食物及药物过敏史;无麻疹、肝炎、结核等传染病接触史。
个人及家族史:G2P2,足月顺产,出生体重3.2kg,出生史正常,生后母乳喂养,生长发育同正常同龄儿,按时进行预防接种。其他家族成员均体健。
入院查体及相关检查:体温36.8℃,脉搏100次/min,呼吸20次/min,血压100/65mmHg,体重26.5kg(同龄儿3%),身高139cm(同龄儿80%)。神志清楚,体态较瘦,呼吸平稳,无鼻翼扇动及三凹征,口周无发绀。颈软,气管居中,胸廓对称,双侧呼吸运动一致,听诊可闻及中细湿啰音。心、腹及神经系统查体未见明显异常。四肢活动自如,无杵状指/趾,肢端温,CRT < 3秒。
入院后完善实验室检查。血常规:C反应蛋白 14mg/L;WBC 13.0×109/L;LY% 20.2%;NE% 74.2%;HB 132.0g/L;BPC 303×109/L;ESR 49mm/h;PCT 0.12ng/ml。炎症指标表明白细胞轻度增高,以中性粒细胞为主,血沉明显增快,结合患儿慢性长期咳脓痰病史,首先考虑细菌感染。结合痰培养及肺泡灌洗液培养均提示铜绿假单胞菌优势生长。胸部CT检查(图1)提示两侧支扩伴感染、纤维灶形成,纵隔淋巴结增大,影像学证实存在支气管扩张表现。铜绿假单胞菌确实为支气管扩张合并感染的主要常见致病菌。根据药敏的结果,予以头孢他啶静滴抗感染治疗,同时予以乙酰半胱氨酸雾化吸入对症处理,辅以拍背、体位引流等物理治疗。同时完善鼻窦CT检查,提示存在全副鼻窦炎,根据耳鼻咽喉科会诊意见,予以局部用药对症处理。结核斑点试验结果阴性。肺功能:最大肺活量1.93L,占预计值的80.6%,正常。用力肺活量1.93L,占预计值的82.9%,正常。一秒量(FEV1)1.25L,占预计值的63.7%,轻度下降。一秒率(FEV1/VCMAX)64.44%,轻度下降。呼气流速各峰值(FEF25/50/75):轻至重度下降。气管镜下纤毛活检电镜检查提示纤毛结构正常。体液免疫检查发现 IgG 15.20g/L,IgA 4.38g/L,IgM 1.43g/L,总IgE 85.40U/ml,均正常;T细胞亚群提示NK细胞25.19%,CD3+% 62.43%,CD3+/CD4+ 29.10%,CD3+/CD8+ 23.70%,CD3-/CD19+11.11%,CD4+/CD8+ 1.23,均在正常范围,常见的体液或细胞免疫功能缺陷依据不足。考虑患儿存在支气管扩张及慢性鼻窦炎,为鉴别Kartagener综合征,明确是否存在内脏反位,予以完善腹部B超检查。腹部B超检查提示存在肝脏弥漫性病变。进一步完善腹部CT提示脾脏稍大,肝脏表面不均匀,门静脉较宽,下腔静脉较宽。心脏超声及肝功能指标正常。综合上述检查及相关病史,患儿既往不存在重症肺炎的病史,无法用单纯的既往感染因素解释目前存在的弥漫性的支气管扩张;实验室检查排除常见的免疫缺陷;影像学检查排除支气管异物;纤毛活检排除原发性纤毛不动综合征;不存在内脏反位,Kartagener综合征的诊断依据不足。但是患儿存在肝脏病变,考虑患儿的支气管扩张存在隐藏的基础疾病,现有的实验室检查无法探寻到病因。根据现有情况,与家长沟通后进行全基因测序检查以进一步明确诊断。经过抗感染治疗,入院第4天患儿咳嗽好转,入院第6天痰量明显减少,入院第12天咳嗽明显减少和肺部啰音消失,予以出院。予以小剂量红霉素,吸入用乙酰半胱氨酸及泛福舒免疫调节剂治疗并临床随访。
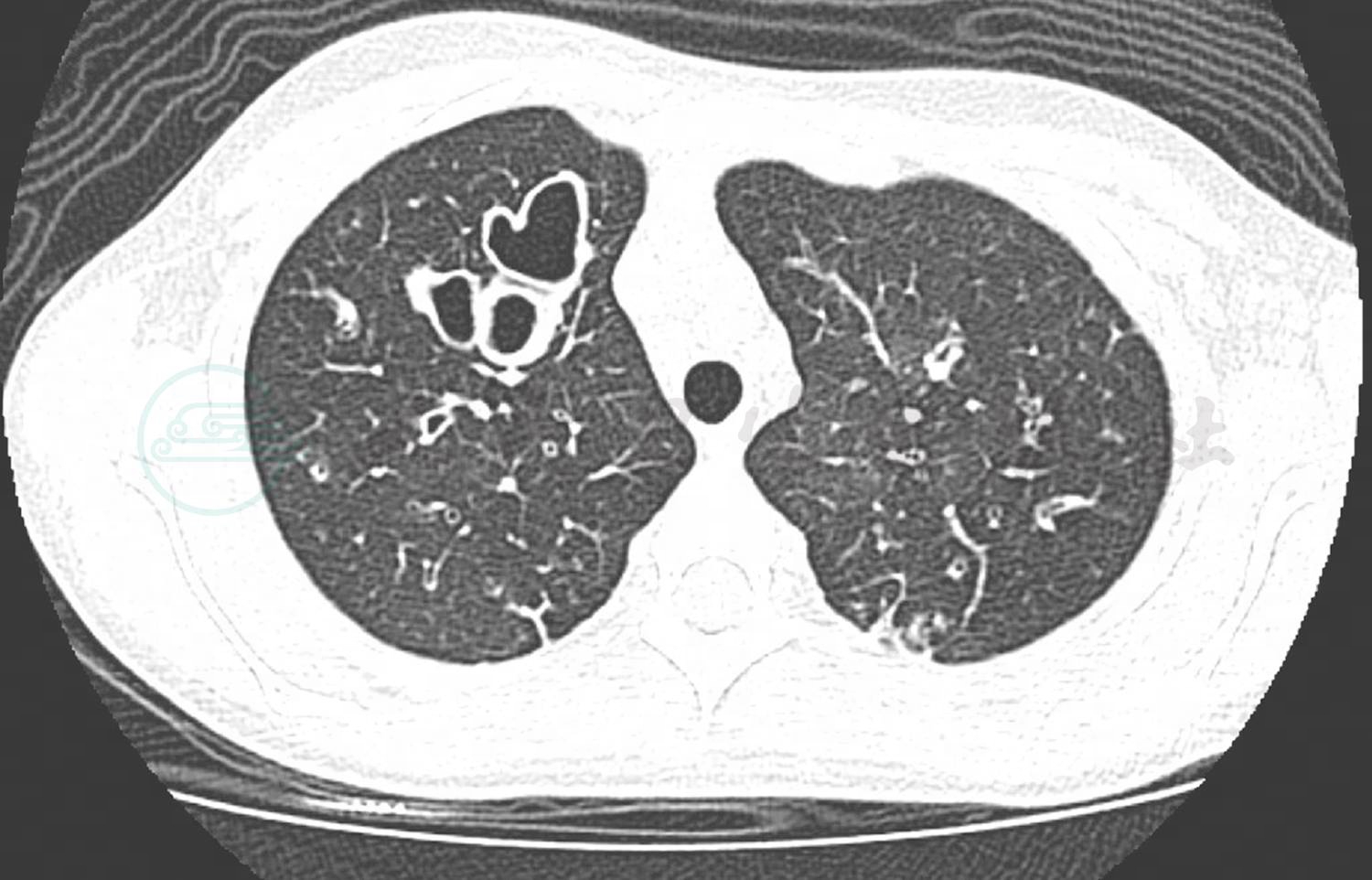
图1 两侧支气管扩张伴感染、纤维灶形成,纵隔淋巴结增大
出院期间3个月患儿仍有晨起咳嗽伴咳黄脓痰,症状时轻时重,期间于当地医院住院抗感染治疗1周左右症状好转出院。期间全基因测序分析结果:CFTR基因存在剪接位点变异c.1766+5G>T(杂合),已有文献报道可导致“囊性纤维化”(图2)和剪接位点变异c.579+1_579+2insACAT(杂合)(图3)。根据患儿的典型的临床表现:慢性咳嗽,支气管扩张,肝脏弥漫性病变需要考虑该患儿为囊性纤维化。CFTR基因检测结果亦支持诊断。
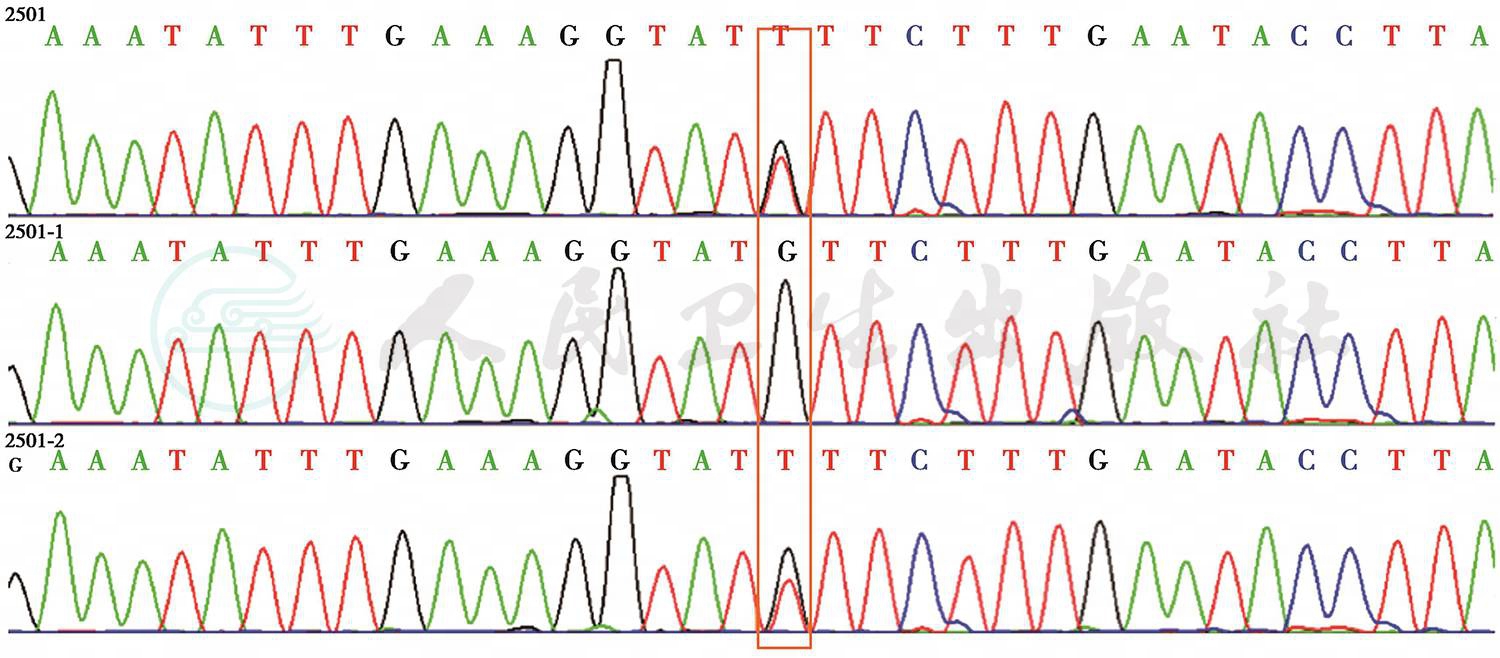
图2 剪接位点变c.1766+5G>T(杂合),由上至下分别为患儿、患儿父亲和患儿母亲基因测序结果
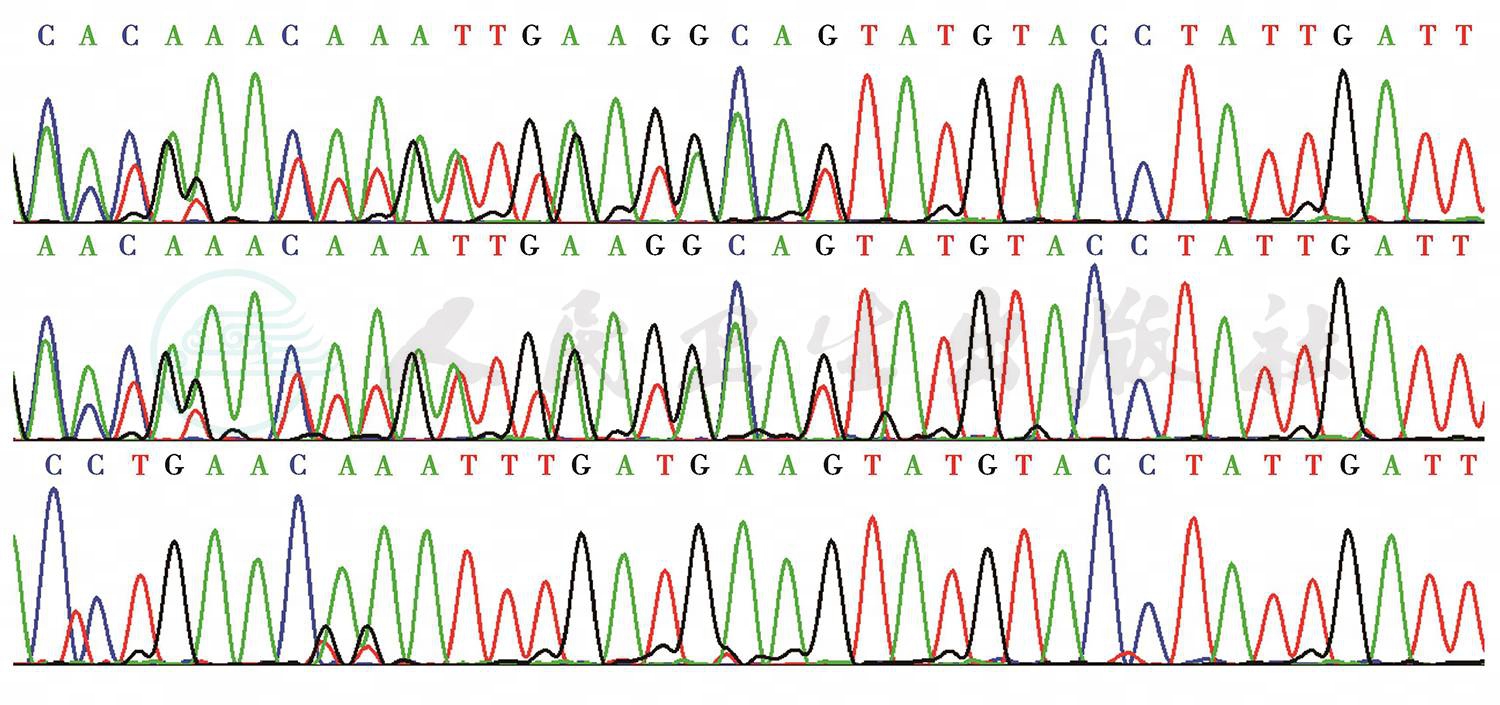
图3 剪接位点变异c.579+1_579+2insACAT(杂合),由上至下分别为患儿、患儿父亲和患儿母亲基因测序结果
出院3个月后,患儿由于咳嗽、咳痰加重伴气促6天,再次至笔者医院治疗。患儿入院时存在明显呼吸困难,鼻翼扇动及三凹征均阳性,未吸氧下SPO2为78%,面罩吸氧下氧合为92%左右,予无创呼吸机辅助通气。痰培养仍提示铜绿假单胞菌,根据药敏予以头孢他啶抗感染,吸痰、雾化等对症处理,同时完善相关检查及脏器评估。入院后患儿有反复发热,予环丙沙星抗感染治疗,4天后体温渐平稳。无创呼吸机通气下患儿呼吸困难渐减轻,入院第7天撤机予面罩吸氧,入院第11天予改用鼻导管吸氧。入院第12天转入呼吸科继续治疗,未吸氧下SPO2维持95%,诉有咳嗽咳痰,双肺可及细湿啰音,活动后气促。患儿病程中定期监测血尿淀粉酶及血脂肪酶均升高,但患儿无腹痛,查体腹稍胀,腹部CT示胰腺饱满伴脂肪浸润可能,加用奥曲肽、胰酶治疗,低脂高蛋白半流质饮食。入院第22天,患儿偶咳少痰,双肺可及少量细湿啰音,未吸氧下氧合可,活动后仍稍有气促,复查肺部CT示肺部炎症吸收,痰栓减少,肺部感染控制可,病情相对稳定,予以出院。出院前肺功能提示FVC占预计值70.1%,轻度下降,FEV1占预计值48.8%,中度下降,一秒率58.44%,中度下降。患儿通气功能异常,存在轻至中度以阻塞为主的混合性病变。
目前乙酰半胱氨酸吸入及肺部物理治疗中,病情较稳定,定期随访肺功能。囊性纤维化作为一种慢病,定期的随访、物理治疗和密切随访至关重要。
囊性纤维化。
诊断依据:①慢性咳嗽,支气管扩张;②肝脏弥漫性病变,胰酶增高;③CFTR基因存在剪接位点变异。
囊性纤维化(cystic fibrosis,CF)是一种侵犯多种脏器的遗传性疾病,主要表现为外分泌腺的功能紊乱,黏液腺增生,分泌液黏稠等。临床上最常影响呼吸道,也有胰脏、肠道、肝脏、肾脏等器官损害。
CF是常染色体隐性遗传疾病,最常见于北欧血统白人,发病率最高的是欧洲、北美和澳大利亚(1/3000新生儿),在南美为1/12000,黑人儿童为1/17000,亚洲人种较为罕见(日本1/350000)。其致病基因是位于染色体7q31的囊性纤维化跨膜传导调节蛋白(CFTR)基因,CFTR是cAMP调节的阴离子通道,在气道、胰道和其他组织上皮细胞的顶端表面表达。当CFTR缺失或活性降低的时候,氯离子和碳酸氢根离子运输也相应地减少,呼吸道黏膜上皮的水、电解质跨膜转运有障碍,黏液腺分泌物中酸性糖蛋白含量增加,改变了黏液流变学的特性,使分泌物变得黏稠,导致管腔阻塞,并最终造成器官损伤。多数囊性纤维化患者于年轻时死亡,但随着近年来治疗的进步,发达国家CF的预期寿命可达50岁,目前患病的美国人可有50%活到成年。
CF的临床表现可为多系统病变,包括呼吸、消化、生长发育及生殖系统等。①呼吸系统:主要表现为反复支气管感染和气道阻塞症状。CF所产生的黏稠黏液阻塞细小气管,并滞留肺部形成细菌繁殖场所,导致肺部反复感染并发生肺部组织的变化。肺部广泛纤维化及肺气肿,有喘息、气促,常并发气胸和纵隔气肿。CF患者鼻窦内的黏液增多也会阻塞鼻窦导致感染,长期反复的鼻窦感染可出现鼻息肉增生,也可能阻塞呼吸道从而引起呼吸困难。②消化系统:新生儿期由于肠道黏稠黏液分泌增加及胰酶缺乏影响蛋白质消化等原因,约10%患儿可出现胎粪梗阻。分泌胰液的胰脏同样也会因分泌物过于黏稠阻塞胰液进入肠道,堆积在胰脏的消化液会引起胰腺炎,并对胰脏产生难以修复的损伤。同时消化道内的胰液不足会引起腹胀、腹泻等消化不良症状并影响营养吸收导致营养不良甚至生长发育不良。由肝脏分泌的胆汁也会过于黏稠阻塞胆管,造成肝脏损害,可出现黄疸。长期可导致肝硬化,出现门静脉高压、食管静脉曲张、脾功能亢进、凝血功能异常等。③内分泌系统及生长发育:当胰脏受损时负责分泌胰岛素的胰岛细胞也会受损死亡,瘢痕胰腺不能产生足够胰岛素,2%~3%的CF患者可发生胰岛素依赖型糖尿病。CF青少年常出现生长缓慢、青春期延迟及活动能力降低等,造成此病症的原因很多,包括长期的肺部感染、营养吸收不良以及过快的新陈代谢率等。④生殖系统及其他:男性与女性均会受到影响。97%男性患者因输精管发育异常,导致无精子或仅有少量精子形成。女性由于子宫颈分泌物过于黏稠,妨碍精子通过,导致生育能力降低。
CF的诊断主要基于临床的典型表现、家族史、证实CFTR失功能的实验室检测或CFTR基因的2处突变。检测方法包括新生儿筛查、汗液氯化物检测以及基因检测等。CF新生儿血液胰蛋白酶水平升高,可在滤纸上采集小滴血液进行新生儿筛查,但此方法不属于CF的确诊性试验。对于非新生儿疑似患者测试时,多进行汗液氯化物检测,通过刺激小范围皮肤发汗,使用离子电渗法收集汗液并进行汗液钠氯含量测定。对于CF患儿汗液氯化物检测诊断标准为汗内氯含量>60mmol/L。除了汗液检测外,通过基因检测CFTR基因是否有突变也可明确诊断。
目前CF尚无可治愈的疗法,主要针对各系统临床表现对症治疗。目前CF治疗药物的发展方向为分子靶向治疗改善CFTR功能,而非单纯局限于下游影响的改善。已有两类药物开始应用或正在发展中。包括CFTR增效剂和CFTR矫正剂。CFTR增效剂能够活化细胞表面已存在的CFTR离子通道,增强离子电流。CFTR矫正剂则通过促进CFTR成熟及蛋白质向细胞膜运输的过程,修复CFTR缺陷蛋白。存在最常见基因突变类型F508del的CF患者,是CFTR矫正剂治疗方法的主要目标群体。
(殷 勇 张 静)









