


 收藏
收藏 已收藏
已收藏患儿,女,11岁。
主诉:活动后气短11年。
现病史:生后6个月即出现活动后气短,表现为呼吸促,不伴口唇及面部发绀,不伴咳嗽及声音嘶哑,喉部无明显“咝咝”声,无活动受限,无呼吸困难。活动后气短症状反复出现,呼吸道感染后加重。家属诉患儿频繁呼吸道感染,每年因呼吸道感染静脉用药次数为3~5次/年。5岁时因反复气短,当地医院诊断为儿童哮喘,间断吸入丙酸氟替卡松吸入气雾剂及硫酸沙丁胺醇吸入气雾剂,用药后症状略有缓解,未坚持规律治疗。近2年家属自觉患儿活动后气短症状较前略减轻,但仍间断出现,为进一步明确诊断来诊。
病来精神状态可,近来无发热、咳嗽,无心悸、胸闷,夜眠无打鼾,无盗汗及体重下降,无皮疹,无吐、泻,饮食及睡眠可,大、小便正常。
既往史:否认异物吸入,无湿疹及鼻炎病史。
过敏及接触史:无食物及药物过敏史。无肝炎、结核等传染病接触史。
个人及家族史:生长发育同同龄儿,按时进行预防接种,无月经初潮,学习成绩不佳。独生子女,父母及一、二级亲属无喘息及哮喘病史,肝炎、结核等家族史。
入院查体及相关检查:神志清楚,精神状态好,下颌稍小,发际线低,呼吸平稳,无鼻翼扇动及三凹征,颈软,胸廓对称,双肺叩诊清音,双肺呼吸音粗糙,听诊偶可闻及呼气相干鸣音,不伴呼气相延长;心、腹及神经系统查体未见明显异常。
由于患儿气促病因不明,且入院时生命体征平稳,无呼吸窘迫及发绀,故入院后未予特殊治疗,仅予完善相关化验检查。
患儿血常规、CRP、肝肾功能心肌酶谱、ESR均正常;体液免疫及细胞免疫指标正常。各项病原学检查肺炎衣原体抗体、结核抗体、呼吸道病毒抗体检测均为阴性;肺炎支原体IgM抗体阴性,IgG抗体阳性。患儿各项生化及病原学检查提示既往支原体感染,无近期感染证据。
为进一步明确哮喘诊断,患儿过敏原及IgE检测均正常;FeNO值在正常范围,行肺通气功能检查提示存在轻度阻塞小通气功能障碍,进一步行支气管舒张试验为阴性,提示该患儿存在气流受限,但经过应用支气管扩张剂后气流受限并未得到改善,因此该患儿表现不符合儿童哮喘的特点。
予患儿行喉镜及喉部CT检查正常,睡眠监测正常,故可除外鼻咽喉部结构异常。肺部CT+三维重建检查提示右肺气管中下段气管狭窄(图1)。因可疑气管狭窄,进而予患儿行局麻下纤维支气管镜检查,术中发现,患儿左肺下叶B6开口缺如,右肺上叶B3及右肺下叶B7开口形成盲端,右侧气管中下段气管轻度狭窄(图2)。因此诊断明确,患儿反复气促为先天性气管发育畸形所致。
此外,心脏三维超声检查未见心血管发育畸形。
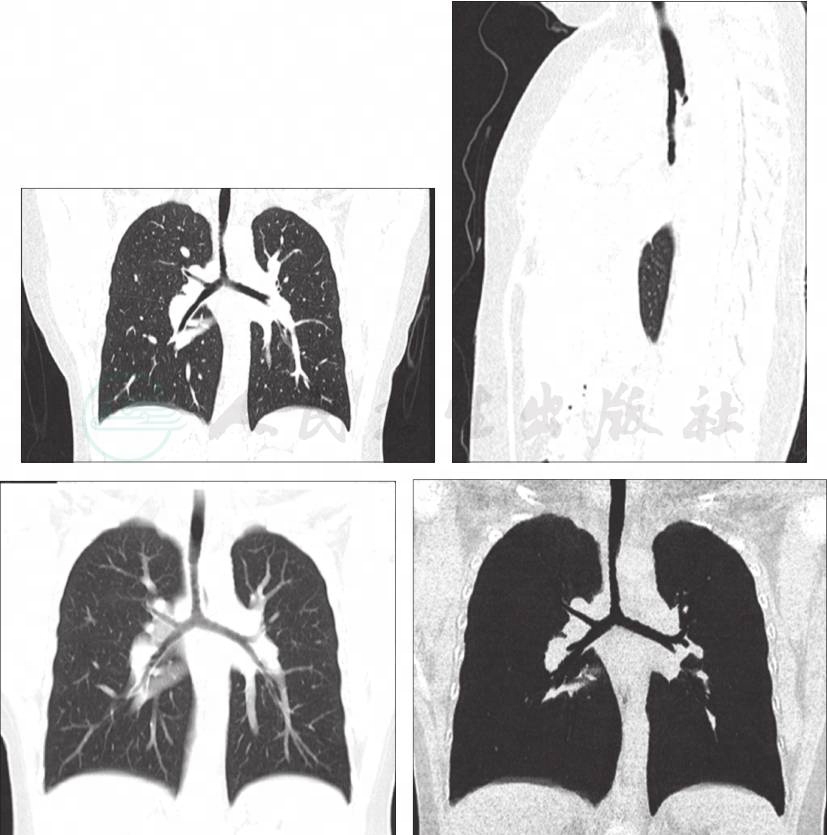
图1 肺部CT+三维重建检查
提示右肺气管中下段气管狭窄
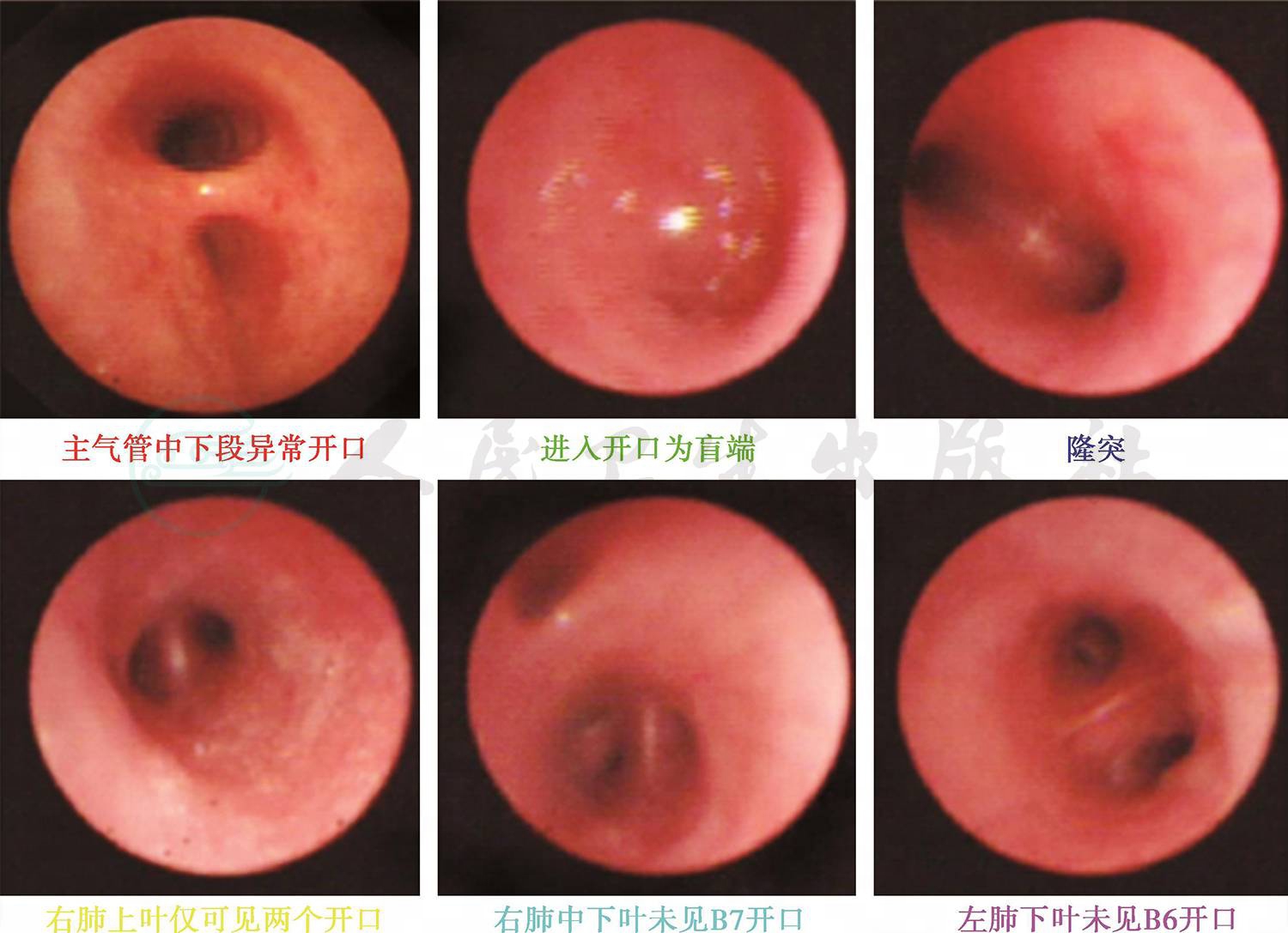
图2 纤维支气管镜下改变
先天性气管发育畸形。
诊断依据:有反复发作的活动后气促症状;发作时肺部查体闻及呼气相干鸣;肺CT三维重建提示右侧气管狭窄,纤维支气管镜检查明确气管狭窄,同时发现气管发育异常,左肺B6开口缺如,右肺上叶B3及右肺下叶B7开口形成盲端;已除外其他心血管疾病、喉部发育异常等。
先天性气管狭窄(congenital tracheal stenosis,CTS)在临床上并不常见,是指由构成气管后壁的完全性气管环和缺少正常结构的膜性气管导致的气管管腔狭窄。其发生可随气管黏膜下层腺体和结缔组织的增生形成管腔,进一步导致阻塞或因血管环压迫以及气管软化等其他原因造成阻塞。
关于CTS的分型有多种分型方法,目前以Cantrell分型和Anton-Pacheco分型应用最为广泛。Cantrell分型依据气管狭窄长度分为弥漫型、漏斗型及节段型。Anton-Pacheco分型依据临床表现:轻,偶有或无临床表现;中,有临床表现,但无呼吸窘迫;重,有呼吸窘迫;A,合并其他畸形;B,无其他畸形。
治疗方面主要以手术治疗为主。1980年前临床医生普遍认为手术治疗CTS极为困难、预后差,通常仅会在抢救时采取气管切开术这样的姑息疗法,只有期望气管自身生长克服先天狭窄以期痊愈,主要以保守治疗为主。随后出现了节段型气管狭窄切除后吻合手术,通常,未超过气管全长1/3的狭窄气管可被安全地切除,而切除超过1/3的狭窄气管,并发症会明显增多,如术后再狭窄、吻合口瘘等。目前所常用的是Slide气管成形术,该术式大大改善了长段以及弥漫型气管狭窄的预后,因为它是完全由自身气管组织扩大管径,不需要其他组织作为补片修补,因此成形术后管壁内有正常气道纤毛及上皮细胞,减少了术后肉芽形成的风险,最终减少了术后再狭窄的概率且术后不需要用气管插管支撑,可以更早拔管,撤离呼吸机,患儿可更早建立自主呼吸。
(刘 芬 尚云晓)









