


 收藏
收藏 已收藏
已收藏一、概述
Larsen综合征最初由Larsen教授于1950年详细描述并命名,表现为特异性的颅面部特征,韧带松弛,多发性关节脱位和足畸形。颅面部特征包括面部扁平且器官相互间距离增大,前额突出,鼻部塌陷。多发关节脱位严重,只能通过矫形手术改善,足部畸形包括马蹄内翻足、马蹄外翻足和“Z”状足。
【发病机制】
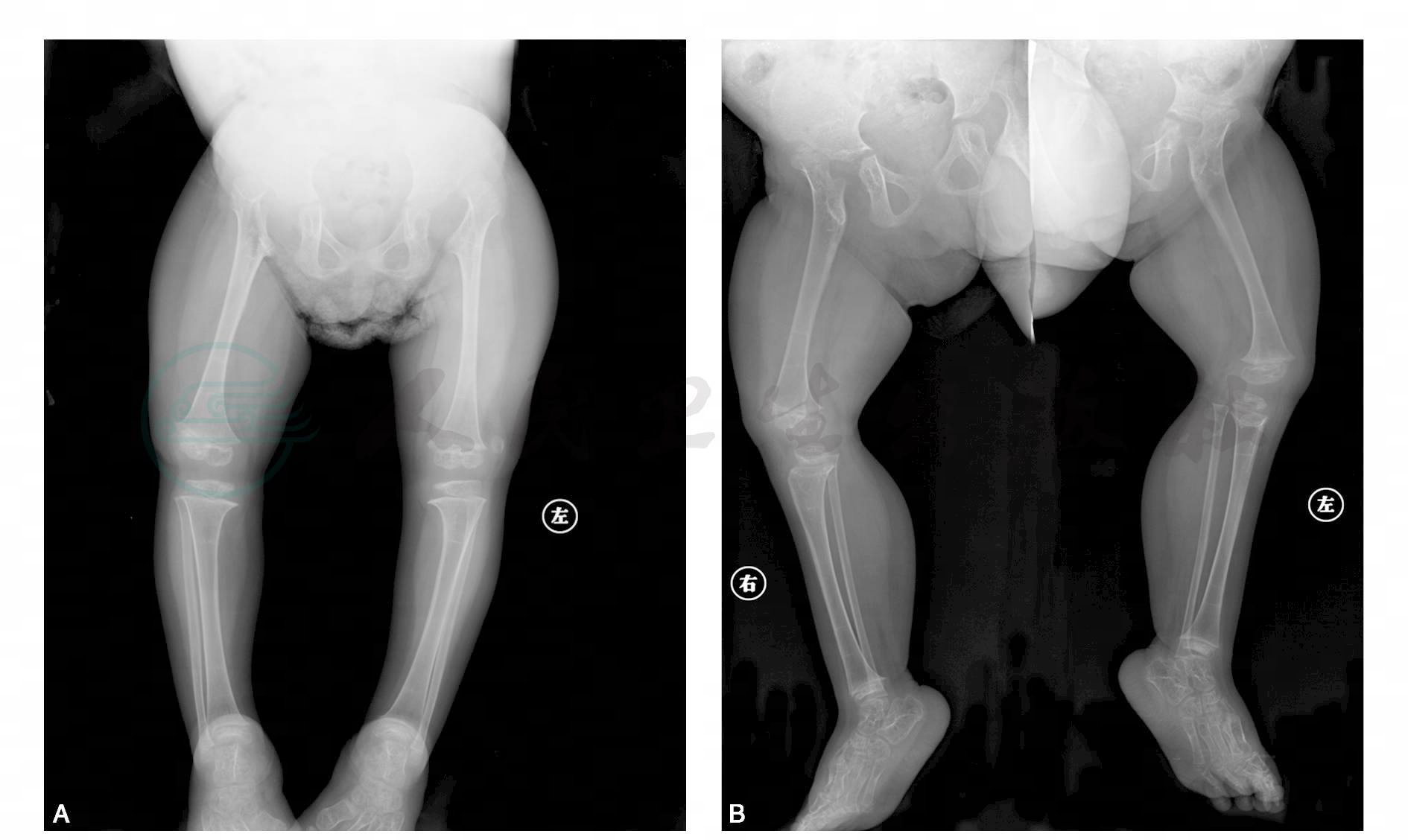
图1 多发骨质密度减低长骨骨皮质变薄,髓腔扩大。四肢长骨明显粗短,干骺端略增宽,骨骺变扁、密度不均,尺桡骨远端及胫腓骨近端关节面倾斜
引自:主编:.疑难病例影像诊断评述.第1版.ISBN:978-7-117-16817-5
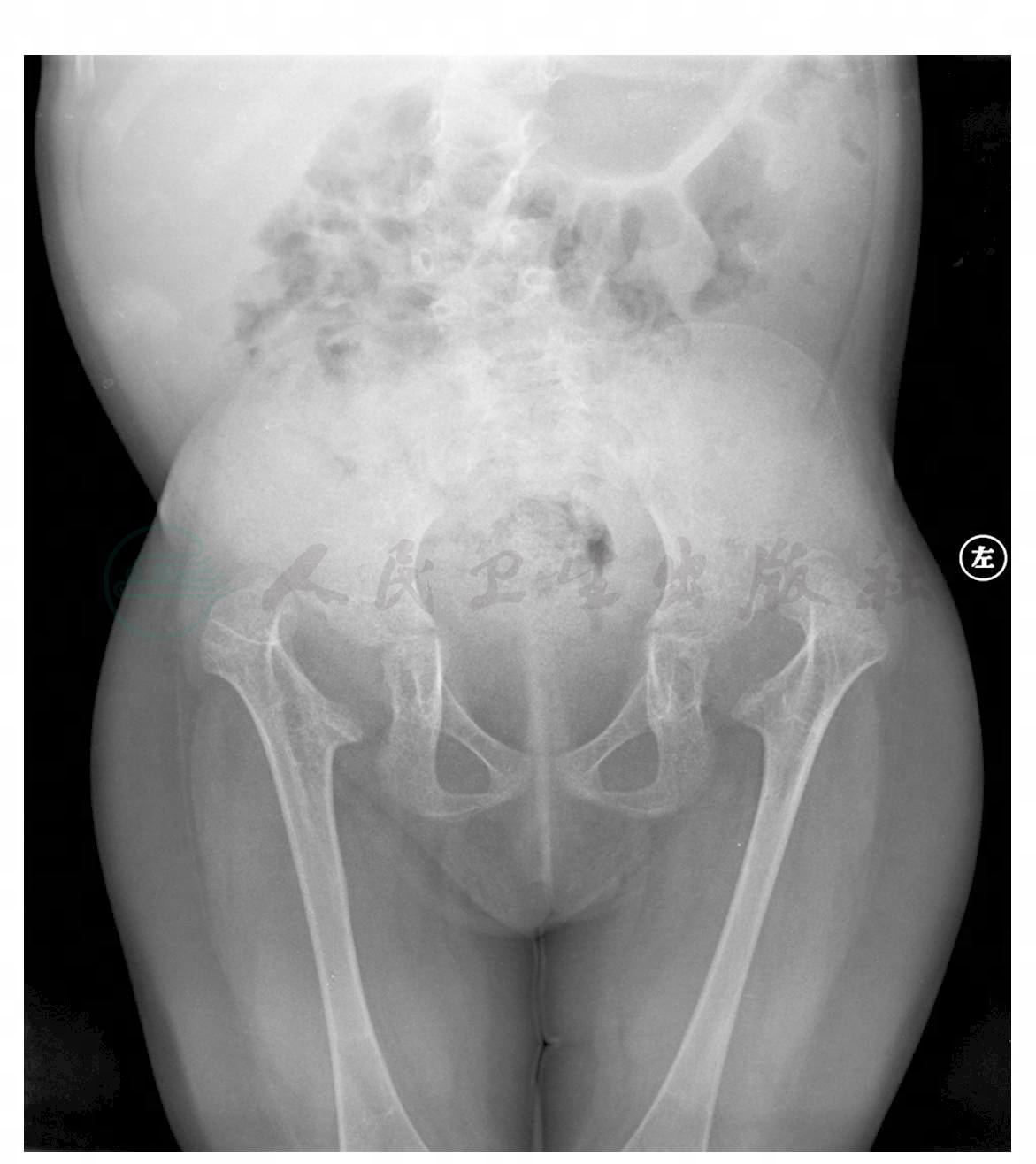
图2 双髋关节、双膝关节及双肩关节脱位,双髋臼发育差
引自:主编:.疑难病例影像诊断评述.第1版.ISBN:978-7-117-16817-5
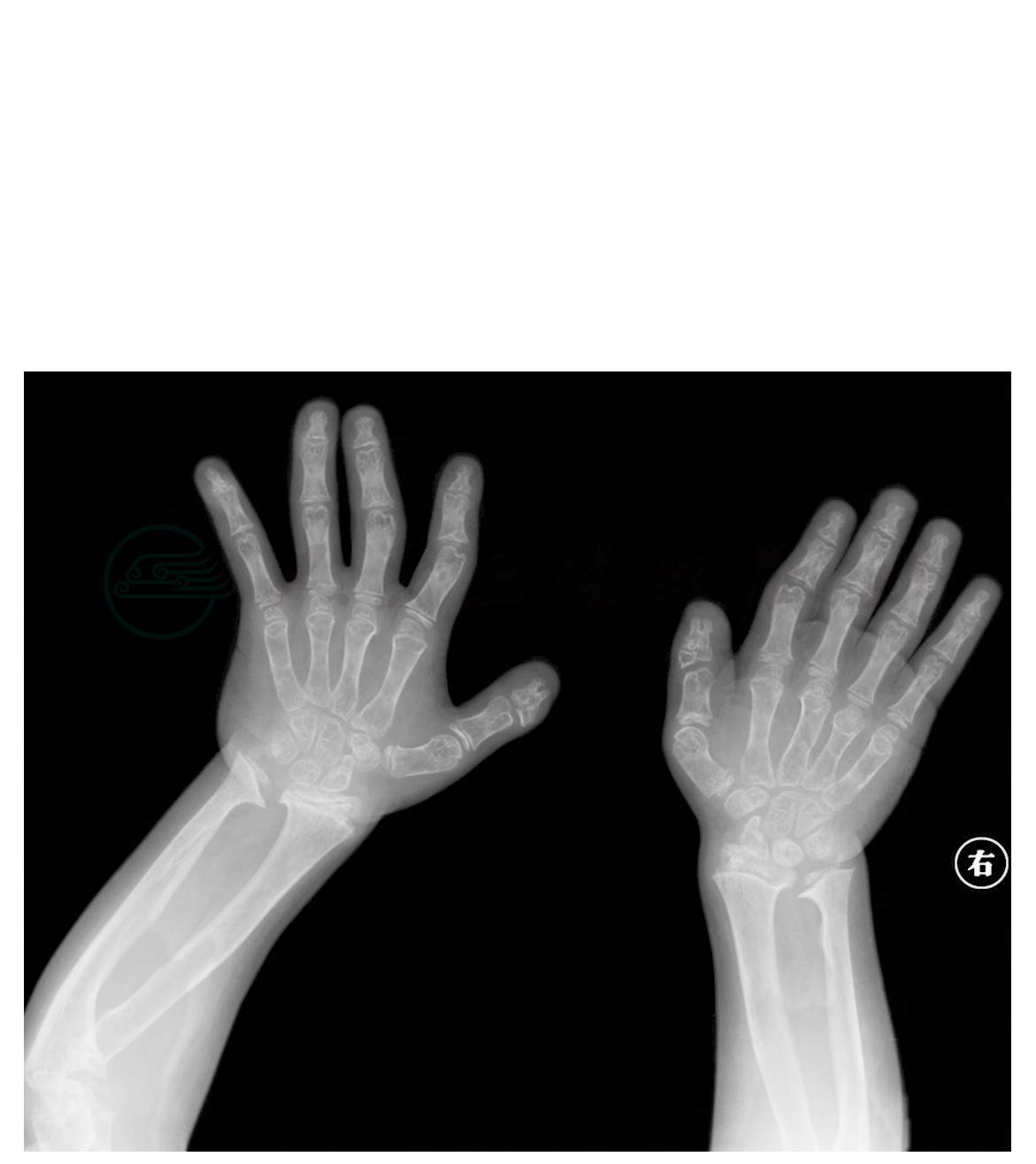
图3 手足多发畸形,第一掌骨及指骨发育短小,远节指骨形态失常、畸形,第2~5近及中节指骨远端弯曲
引自:主编:.疑难病例影像诊断评述.第1版.ISBN:978-7-117-16817-5
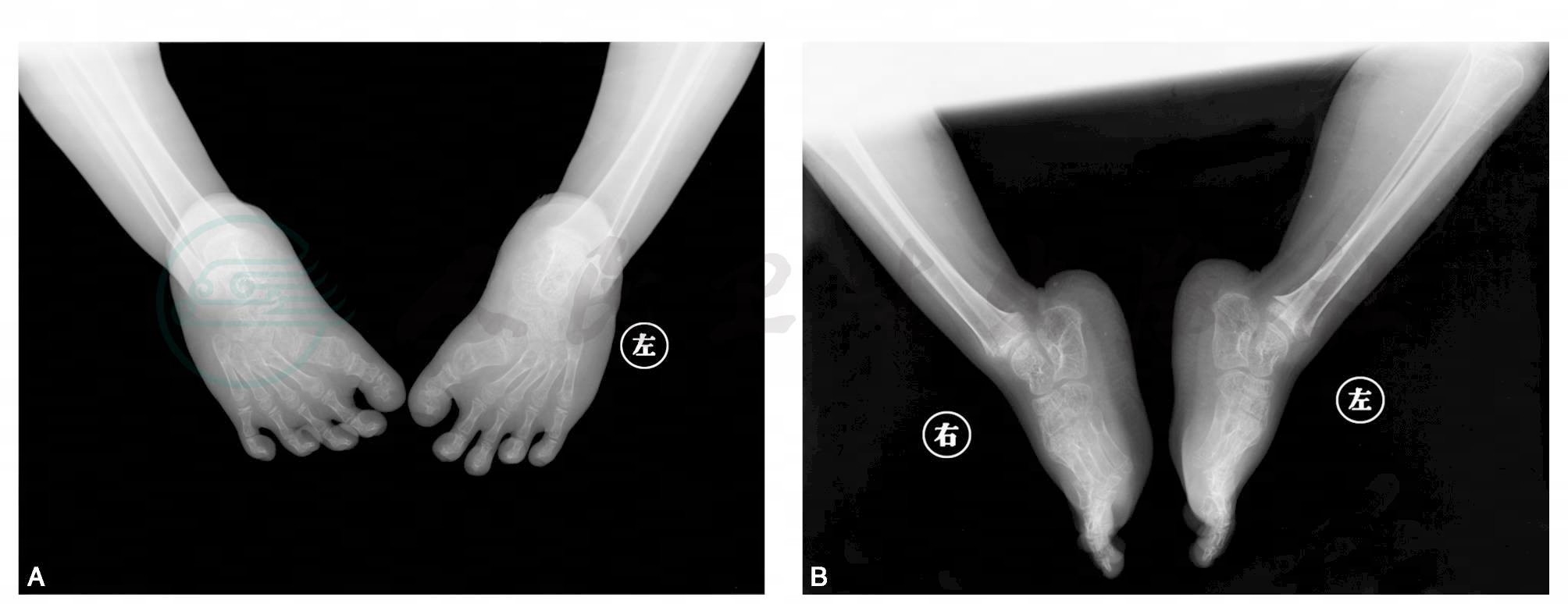
图4 双足内翻,距骨轴线偏离第一跖骨轴线,跟骨轴线偏离第四跖骨轴线,跟距角减小,双足第一跖骨增粗膨大
引自:主编:.疑难病例影像诊断评述.第1版.ISBN:978-7-117-16817-5
Larsen综合征的发病率约1/100000,属于罕见病。目前认为其病因为连接组织和胶原组织存在遗传缺陷,导致全身结缔组织发育障碍,引起关节过度松弛而发生脱位、颜面改变、手足变形、身材矮小以及心脏、脊柱畸形。该病是常染色体显性遗传性疾病,在近亲结婚夫妇的该病患儿表现为常染色体隐性遗传。常染色体显性遗传基因位于3p14染色体,该染色体区域存在丝蛋白B编码基因,该基因中结构功能域的改变可引起骨骼缺乏或发育不全、骨化障碍、脊柱发育异常、关节脱位和矮身材。除遗传性基因缺陷之外,Larsen综合征患儿本身的丝蛋白B基因发生突变,也是Larsen综合征的发病原因。
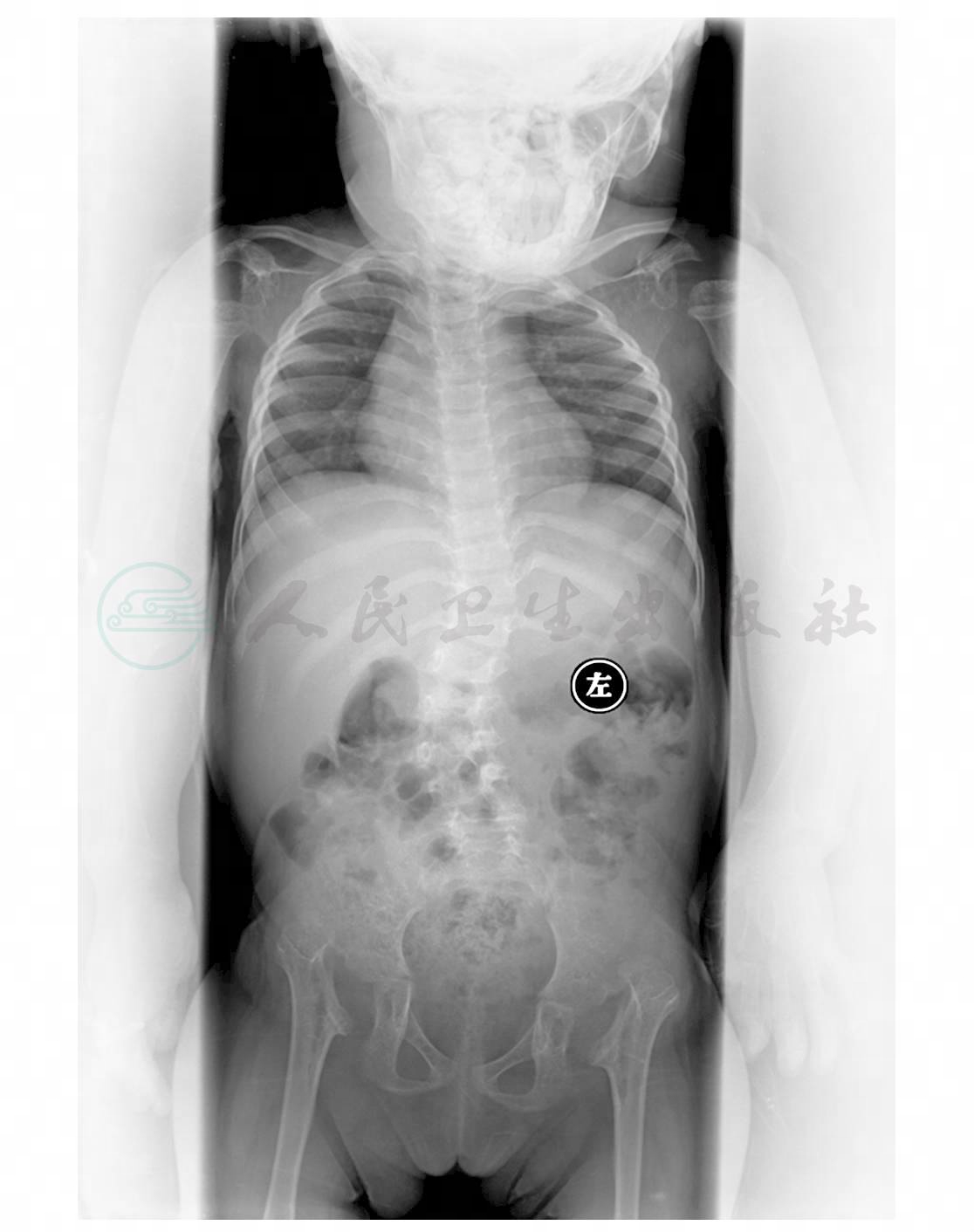
图5 胸腰段脊柱以第2腰椎为中心向右侧弯曲,Cobbs角约38°
引自:主编:.疑难病例影像诊断评述.第1版.ISBN:978-7-117-16817-5
【临床表现】
Larsen综合征的临床症状和病情严重性不同患儿间差异较大。Larsen等初次报道的病例包含了多发的先天性的关节脱位(通常是髋关节、膝关节、肘关节)合并颜面部特征性改变(额部隆起,鼻部塌陷,面部扁平及器官距离增宽),手指铲状畸形,跟骨双骨化中心,脊柱异常常导致脊柱稳定性降低甚至脊髓损伤。虽然相对于多发关节脱位和颜面部改变,脊柱异常的发生率较低,主要表现为脊柱侧弯和颈椎后突畸形,但是由于颈椎后突易压迫脊髓引起脊髓损伤危及生命,所以脊柱异常在临床诊治过程中需格外重视。脊柱侧弯多发生在胸腰椎,颈椎后突多以下段颈椎后突明显,引起脊柱侧弯和颈椎后突的原因是椎体发育不良。Larsen综合征的治疗原则是分步骤、分阶段外科矫形手术治疗。外科矫形治疗过程遵循先纠正脊柱异常,后纠正关节异常,如有需要最后进行颜面部整形治疗。由于该病的病床表现和严重程度差异很大,临床治疗的效果不尽相同。
Larsen综合征也可出现心脏异常,类似于马方综合征。心脏改变分先天性和后天性两类。先天性异常改变包括房间隔缺损、室间隔缺损和动脉导管未闭;后天性异常改变包括主动脉扩张/功能不全、二尖瓣脱垂/功能不全、动脉瘤。Larsen综合征可伴有呼吸系统异常,主要包括喉软骨软化病、气管软化、支气管软化、声门下狭窄。
二、鉴别诊断
Larsen综合征需与爱唐综合征、软骨发育不全、耳腭指综合征、黏多糖病、马方综合征相鉴别。
1.皮肤弹力过度症(cutis hyperelastica)
也称爱唐综合征(Ehlers-danlos syndrome,EDS),是一种原因尚未完全明了的多为常染色体显性遗传性疾患,也有隐性遗传者或伴性遗传者。EDS特征为患儿皮肤弹力过度,拉长后立即弹回,如将颈部、肘部或其他较松的皮肤向外拉出,可以距离很长而且立刻回缩。其次是皮肤脆弱,表现在面部及四肢的皮肤血管很脆弱,受微伤后容易发生皮肤瘀斑或皮下血肿,损伤大血管可致大出血,以后形成萎缩性紫色瘢痕。有时齿龈在刷牙后即易出血。偶可发生鼻出血、便血、咯血。如皮肤发生裂伤,伤口缝合后,易扯裂且缓慢愈合,最后形成范围较大的薄膜状萎缩性瘢痕,间有微小皱纹。还有关节活动过度,表现关节松弛,特别以手指过度伸直较为多见,拇指可以向后弯曲,与腕部接触。由于体内结缔组织都软弱无力,常并发脐疝、腹股沟疝、膈疝及膈膨升还可与其他先天性疾病如先天愚型、先天性肌肉骨骼畸形、蜘蛛脚样指、先天性成骨不全等同时发生。基于临床表现和分子生物学缺陷,目前分11种亚型,但尚不能包罗本病。
Larsen综合征以面部特征及多发关节脱位表现为主,皮肤弹性差为次,而爱唐综合征临床表现虽可合并肌肉骨骼畸形、蜘蛛脚样指、先天性成骨不全及髋关节脱位,但其特征性的临床表现为以皮肤弹力过度,可以与Larsen综合征相鉴别。另外,凝血试验检查结果常提示出、凝血时间延长,束臂试验阳性,但血小板计数正常,也可提供有价值的鉴别诊断信息。
2.软骨发育不全(achondroplasia,ACH)
是一种常见的软骨发育不良,又称胎儿型软骨营养障碍( chondrodystrophia fetal)、软骨营养障碍性侏儒(chondrodystrophic dwarfism)等。这是一种由于软骨内骨化缺陷而导致的发育异常,临床上以四肢短、躯干相对正常、巨头、脊柱胸腰段后凸、椎管狭窄为特征。ACH的发病机制与成纤维细胞生长因子受体-3(FGFR-3)基因跨膜区的点突变密切相关,80%~90%的病例是散发的,为新生突变。本病为先天性发育异常,有明显的遗传性及家族史,为常染色体显性遗传。如父母一方有病,子女中1/2可以得病;如父母均为患者,则子女几乎都要受累。由于不少病人不结婚或难产,致使无下一代,因而影响到遗传形式。所以散发性病例占90%。当然也有人是由于基因突变所致。在双胎中可以1个患病,亦可以2个均有,女性略多于男性。该病的基本病理改变发生在软骨化骨过程,长骨纵向生长受阻,而膜内化骨过程不受影响,故骨的粗细正常,但因长度减短而相对变粗。骨骺软骨细胞可发生及增殖,但不能进行正常的钙化与骨化,因而骨端增大。镜下可见软骨细胞不能像正常那样呈规则的柱状排列,而是分散,不规则成堆,骨化过程的多个区域,如静止区、增殖区、肥大及预备钙化区等的层次也发生紊乱,干骺端毛细血管不能有规则地进入骺进行正常的吸收,成熟的软骨细胞不能钙化,影响了骨的生长。另外,广泛的软骨黏液样变性,细胞肿胀,细胞核增大,基质呈半流体结构,病变部位的软骨骨化延迟,呈斑块状分布,而斑块间的钙化过程则比较正常。
ACH患者特征性影像学变现为脊柱畸形,在X线片上的主要表现包括椎体小且伴有椎弓根短及腰椎椎弓根间距窄。腰椎管狭窄,腰椎管横径(即椎弓根间距)从L5到L1逐渐变小,而正常情况是逐渐增大;腰椎体不规则变扁,后缘凹陷;骶椎发育较小,后翘呈水平位;腰骶角增大。其他畸形颅底和面骨发育障碍,面骨发育小,颅面比例加大,前额突出,下颌前突;骨盆前后径明显小于横径,呈扁平骨盆;髋臼缘不规则,髋臼平;髋臼角明显变小(常<10°);股骨颈粗短,股骨头骨骺核出现晚;长管状骨骨干短粗,髓腔变窄,干骺端增宽,以股骨远端、胫骨近端最显著;掌指骨粗短,同排骨近于等长,指不能并拢,呈“车轮状”或“三叉戟”。ACH与Larsen综合征均具有鲜明的影像学表现,但明确鉴别仍需注意以下几点:
(1)二者都可变现为长骨粗短,但ACH长骨变短更明显,其增粗是相对于长骨不能正常增长而出现的,而Larsen综合征长骨无论是增长还是增粗均呈发育障碍表现。
(2)ACH骨骺发育延迟、出现晚,Larsen综合征骨骺出现的时间并不晚,只是关节松弛脱位等因素影响其进一步发育,从而表现的仅是形态上的失常。
(3)ACH髋关节改变表现为扁平髋,而Larsen综合征表现为髋关节脱位。
3.耳腭指综合征(otopalatodigital syndrome)
1962年由Taybi首先报道,又名Taybi综合征,主要表现为传导性耳聋、腭裂及骨骼(指趾常见)结构异常。主要的临床体征:身材矮小,轻度智力低下。中度传导性耳聋,腭裂。前额隆凸,枕部隆凸。面骨发育不良,眼距宽,外侧眶上嵴丰满,鼻小,鼻根宽,嘴小。萌牙延迟,部分牙缺如。躯干小,漏斗胸。肘关节伸展或旋前受限,拇指(趾)远端指(趾)骨短宽,指(趾)甲短,偶有髋关节脱位,前囟闭合延迟,并趾等。与Larsen综合征相同点:①身材矮小;②前额隆凸;③面骨发育不良,眼距宽,鼻小。如下几点可与Larsen综合征相鉴别:
(1)关节受累主要体现在肘关节伸展或旋前受限,偶有髋关节脱位;而Larsen综合征表现为明显的多发关节脱位。
(2)耳腭指综合征拇指(趾)远端指(趾)骨短宽,指(趾)甲短,并趾畸形,而Larsen综合征手足部的改变由于结缔组织发育障碍表现为形态失常、变形为主,如马蹄内翻足、马蹄外翻足和“Z”状足,指骨增长、弯曲等。
(3)耳腭指综合征有明显的听力障碍甚至耳聋,可伴有智力低下; Larsen综合征听力及智力正常。
4.黏多糖病( mucopolysaccimridosis,MPS)
又称黏多糖沉积症,在黏多糖降解过程中,溶酶体内先天性缺乏某些水解酶(硫酸酯酶或糖苷酶)致使黏多糖不能分解而沉积于全身组织引起疾病。MPS是一类遗传性代谢性疾病,根据不同酶的缺陷分为8型,Ⅰ型和Ⅵ型相对常见,其余各型临床罕见,除Ⅱ型为X性连锁隐性遗传外,其他均Y染色体隐性遗传,故常无阳性家族史。MPS骨骼系统的X线表现:
(1)头颅增大呈舟状,蝶鞍变扁增宽,似横置的小提琴状,蝶窦及乳突气化不良,板障增宽或局限性内板增厚,眶顶和颅底骨致密硬化,部分患儿前额明显突起。
(2)腰椎生理弧度异常,椎体前缘呈鸟嘴状突出,常伴有后突畸形。
(3)胸骨前突形成鸡胸,肋骨前部飘带样增宽,胸骨分节,柄、体、剑突均不融合。
(4)爪形手,双手指骨粗短,近节指骨远端变尖,指骨基底部、掌骨远端干骺端增宽、凹陷,如“爪形”。
(5)四肢长骨骨干塑形障碍,变得粗短,尺桡骨远端关节面相互倾斜,四肢长骨的改变被认为最有诊断价值。
(6)骨盆变形,髋关节间隙和耻骨联合增宽,股骨头发育不良。
本病与Larsen综合征在影像学鉴别上比较容易,二者分别具有特征性的X线表现,MPS胸骨及肋骨的改变比较典型,而Larsen综合征胸部改变不明显,主要为多发关节脱位;本病与Larsen综合征长骨改变均粗短,但长骨干骺端改变不同是二者较明显的鉴别点;二者额面部特征可表现为额部前突、鼻部塌陷,但MPS另有蝶鞍形态改变。临床表现均为患儿身材矮小,Larsen综合征智力正常,而MPS患儿智力低下。
5.马方综合征
是一种遗传性多系统的结缔组织病,由原纤维蛋白-1因子(FBN-1)突变引起。该综合征具有广泛的临床表现。心血管表现常见,也可并发脊柱侧凸、漏斗胸、蜘蛛指(趾)和髋臼突出等骨骼畸形表现。脊柱侧凸是马方综合征常见和潜在的严重表现,可以导致心肺损害和使肺容量受限。大约66%的马方综合征患者既有漏斗胸亦有鸡胸,由于肋骨纵向生长过快造成。蜘蛛指是马方综合征的特征性骨骼改变,髋臼突出发生率很少,大约在16%~27%。马方综合征扁平足的发生率大约25%,表现为足部扁平外翻,目前认为是由韧带松弛引起。
由于均可引起骨骼及心血管系统改变,因此,Larsen综合征需与马方综合征进行鉴别。二者相同点是均可出现脊柱侧凸、足部外翻等畸形,不同点是,Larsen综合征的特征性改变在于明显多发关节脱位和颅面部改变,而马方综合征的特征性改变在于胸骨改变和手指骨改变。
本例患儿来我院骨外科就诊,拟在我院行脊柱侧弯矫正术,在拍摄脊柱片和髋关节片时发现骨骼和髋关节X线有异常改变,于是完善补充摄片部位并查看患儿临床体征,由于诊断经验有限,加上Larsen综合征属罕见病,初次诊断并没有明确诊断为Larsen综合征。经查阅学习文献,结合患儿临床表现后作出了Larsen综合征的影像学诊断。该患儿后经全院会诊并进一步完善临床各项相关检查,最终明确诊断为Larsen综合征。通过复习本病例及相关文献,可以明确Larsen综合征的影像学改变特点,虽然该病罕见,但本病例具有典型的临床表现和影像学特征,有助于拓宽放射科医生的知识面及提高放射诊断水平。
【病例介绍】
女,3岁,因运动发育异常近3年就诊。体格检查:身材矮小,不能站立,发育落后,营养中等,智力一般,全身皮肤弹性明显差,面容特殊,双眼距增宽,双巩膜蓝染,鼻梁塌陷,鼻孔大略外翻,头围46cm,囟门已闭;听力正常,甲状腺无肿大。胸廓对称,胸围45cm,肋缘外翻,双肺呼吸音清,心音有力律齐,未闻及病理性杂音。全腹平软,肝肋下2cm。胸腰段脊柱以第2腰椎为中心向右侧弯曲,四肢关节松弛,肌张力弱,肌力2~3级,双髋关节外展(+),双髌骨外移,双膝不稳定,双足扁平(图1~图5)。
【诊断】
Larsen综合征









