


 收藏
收藏 已收藏
已收藏患者,女性,20岁,广东省高州人。
病史
主诉
四肢进行性乏力、气促10余年,加重1月余。
现病史
患者于10多年前无明显诱因出现四肢乏力,活动后气促,运动耐量逐渐降低,不能进行短跑等体育活动,当时未予以重视。之后病情呈进行性加重,逐渐发展至完全不能参加体育活动。1年前开始出现日常活动后乏力、气促及吞咽无力,但进食尚可,可站立、行走、持物,无肌肉酸痛、发热。逐渐出现全身消瘦,至今体重下降约10余斤。患者1月前出现乏力、气促明显加重,伴心悸,活动后尤甚,至深圳市人民医院、高州市人民医院就诊。胸片示“肺血流减少,肺动脉稍隆凸”;胸部CT示“心影稍增大,左室增大为主,肺动脉段增粗、膨隆,右侧少量胸腔积液,心包积液”;右心导管检查示“肺动脉压42/33mmHg,右室压49/13mmHg”,拟诊“原发性肺动脉高压”住院治疗。经降低肺动脉压、改善心功能等治疗后,症状无明显好转。于半月前气促加重,并出现呼吸困难,血气分析拟诊为“Ⅱ型呼吸衰竭”,予以呼吸机辅助呼吸,并转神经内科治疗。体检肌
-张力偏低,双上肢肌力5级,双下肢肌力3级,腱反射减弱,病理征阴性,拟诊为“肌无力查因:线粒体肌病?”,予以细胞能量、神经营养、B族维生素及对症支持治疗后,症状无明显好转,不能脱机,4天前行气管切开术,为进一步诊治转入我院。起病以来,患者无肢体麻木,无肌跳,无视力下降、听力下降。
既往史
幼时体弱,常患“感冒”,治疗后可好转;否认肝炎、结核病史,无中毒、外伤史。
过敏史
未发现。
婚育史
未婚育。
个人史
生长发育较同龄人延迟,身材较矮小,学习成绩较差。
家族史
家族中未发现有类似疾病者及特殊遗传病史。
体格检查
一般情况
体温36.5℃,脉搏72次/分,呼吸16次/分(呼吸机辅助呼吸),血压110/70mmHg;发育、营养较差,神志清楚,精神可,被动体位,查体合作。
专科情况
精神智能状态
无幻觉、妄想,时间、地点、人物定向力正常,近、远事物记忆力及计算力正常,无失读、失写,理解力正常,无失语、失认及体象障碍。
脑神经
嗅觉及视力粗测无异常,双侧视野手试法未见缺损;双眼底视乳头边缘清,A∶V=2∶3,未见动静脉压迹,视网膜未见出血、渗出。无眼裂增大或变窄,双眼睑无下垂,双眼球居中,无突出、凹陷,眼球各向运动正常,无凝视,无复视,双侧瞳孔等圆等大,直径约3.5mm,双侧直接、间接对光反射灵敏,调节、辐辏反射正常引出。双侧面部痛、触觉正常对称,温度觉未查,双侧咀嚼肌无萎缩,咀嚼动作对称有力,张口下颌不偏,双侧角膜反射正常,吸吮反射、下颌反射未引出。能闭目、鼓腮、露齿,双侧鼻唇沟对称,口角无偏歪;听力粗测及音叉试验无异常;无声音嘶哑、构音障碍、饮水呛咳、吞咽困难,双侧咽反射存在。双侧胸锁乳突肌与斜方肌饱满,双侧转颈、耸肩自如。伸舌居中,无舌肌萎缩及震颤。
运动系统
右利手,四肢肌肉明显萎缩;四肢肌张力降低;双上肢近端肌力3级,远端5-级,双下肢近端肌力2+级,远端3级;未见共济失调及不自主运动。
感觉系统
双侧痛觉、触觉、温度觉对称存在,双侧音叉振动觉未查,关节位置觉、运动觉正常,双侧形体觉、定位觉、两点辨别觉及图形觉正常。
反射
双侧肱二头肌反射(+),双侧肱三头肌反射(±),双侧桡骨膜反射(-),双侧膝反射(-),双侧踝反射(-)。
病理反射
未引出。
脑膜刺激征
未引出。
自主神经系统
全身皮肤温度正常,湿度适中,弹性好,毛发、指(趾)甲营养好,排尿、排便正常,皮肤划痕试验阴性。
辅助检查
实验室检查
血液学
(我院)空腹血糖正常。肌酶学检查:肌酸激酶(CK) 321U/L 、肌酸激酶同工酶(CK-MB) 31U/L、丙氨酸氨基转移酶(ALT) 51U/L、天门冬氨酸氨基转氨酶(AST)57U/L 、乳酸脱氢酶(LDH) 249U/L、血沉32mm/h、血乳酸2.8mmol/L。
影像学检查
胸片
示两肺条索状改变。
心脏彩超
左心室室壁稍厚,收缩功能正常。
电生理学检查
肌电图
右侧肱二头肌、右股直肌、左胫前肌肌源性损害。
病理学检查
肌肉活检(图1~图5)。
光镜与电镜诊断意见:成人Ⅱ型糖原累积病,病变严重。
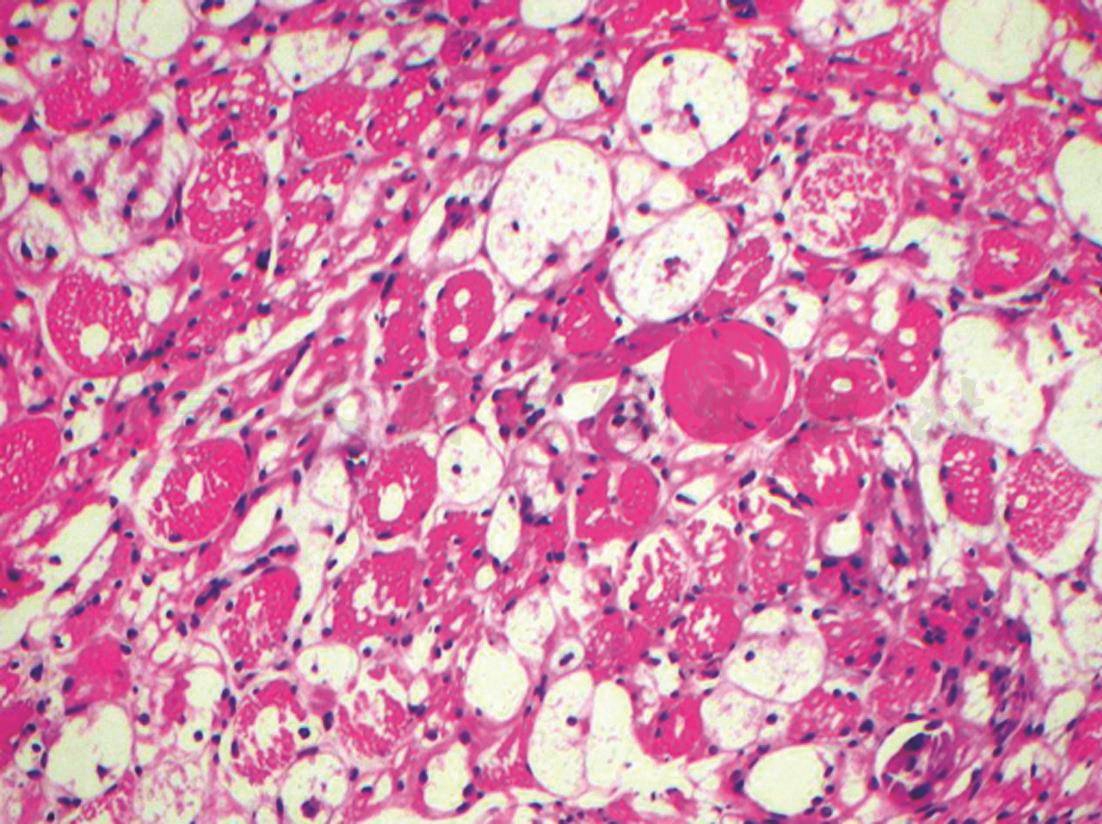
图1 肌活检(光镜下HE染色)
肌纤维肌浆广泛溶解,多数肌纤维呈空泡状,仅见肌膜存在,残留的肌纤维有大小不等的空泡,肌束中血管内皮细胞增生,管腔狭窄
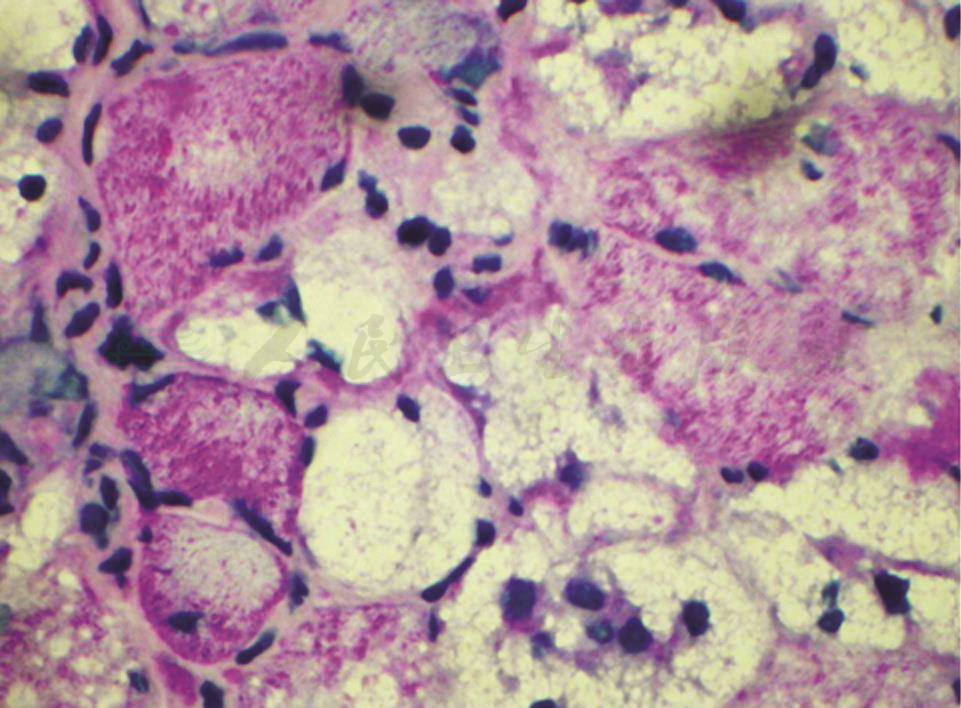
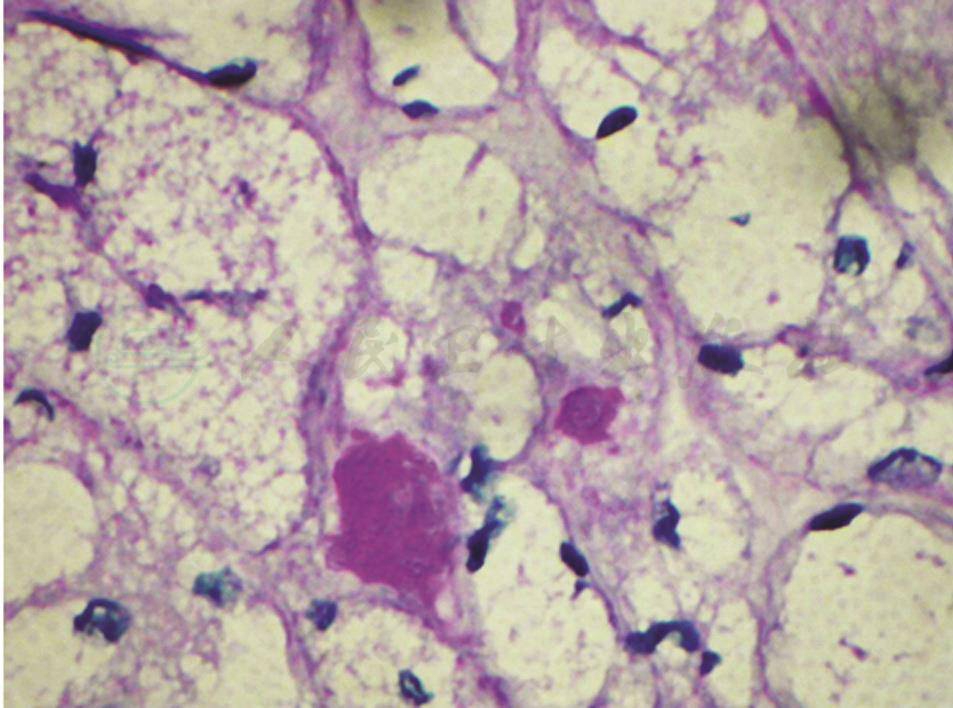
图2 肌活检(光镜下PAS染色)
显示严重空泡变性的肌纤维不着色,但少数肌纤维见明显阳性(箭头所指),提示有糖原累积
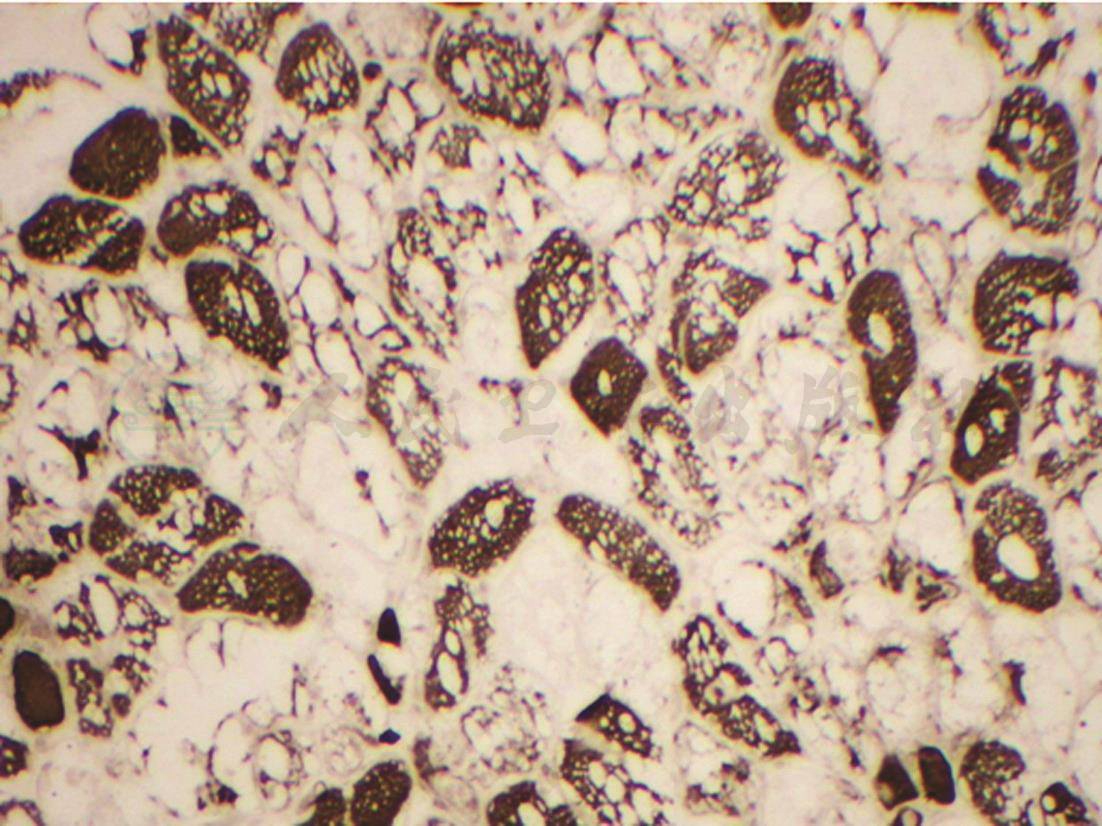
图3 肌活检(光镜下ATP酶染色)
肌纤维比例和分布基本正常,空泡变性的肌纤维主要以Ⅰ型纤维为主。NADH-TR染色未见轴突改变,改良的Gomori染色未见碎红纤维,油红 染色未见脂肪沉积
染色未见脂肪沉积
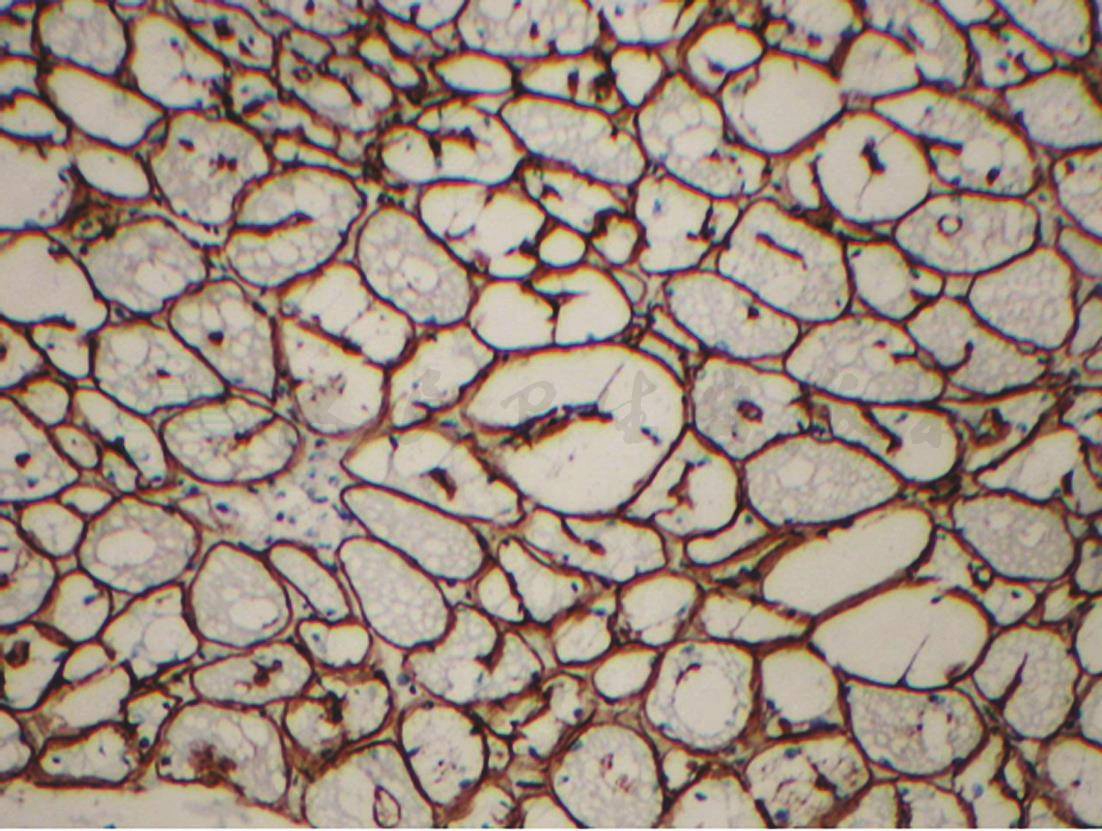
图4 肌活检(光镜下组织化学染色)
Dystrophin免疫组化(+)
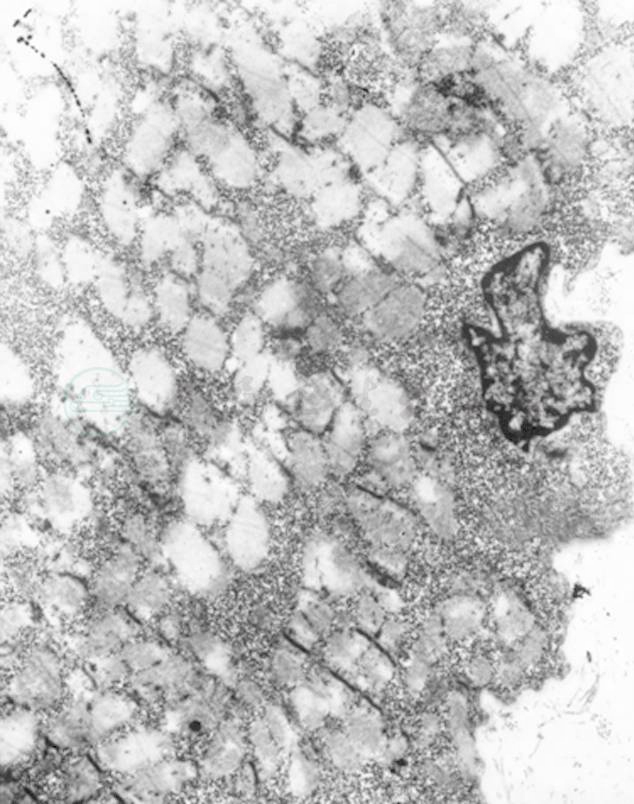
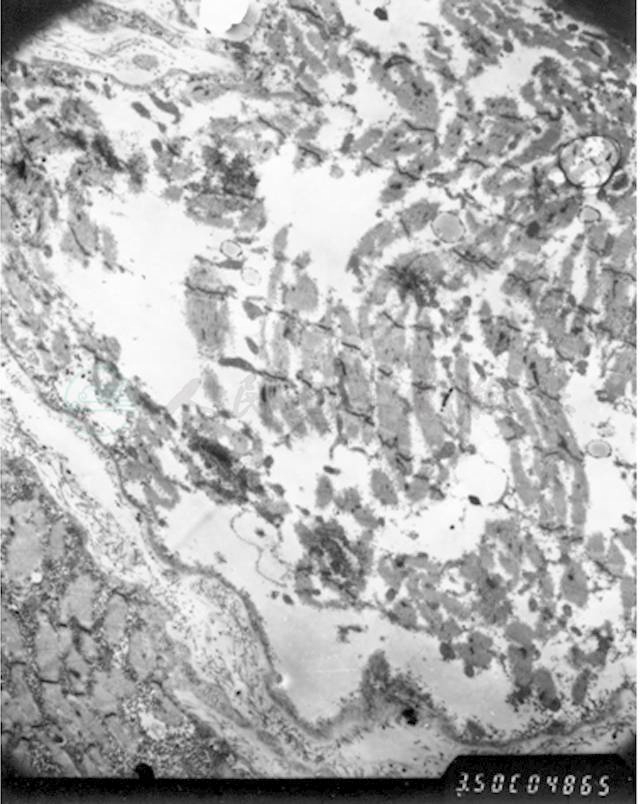
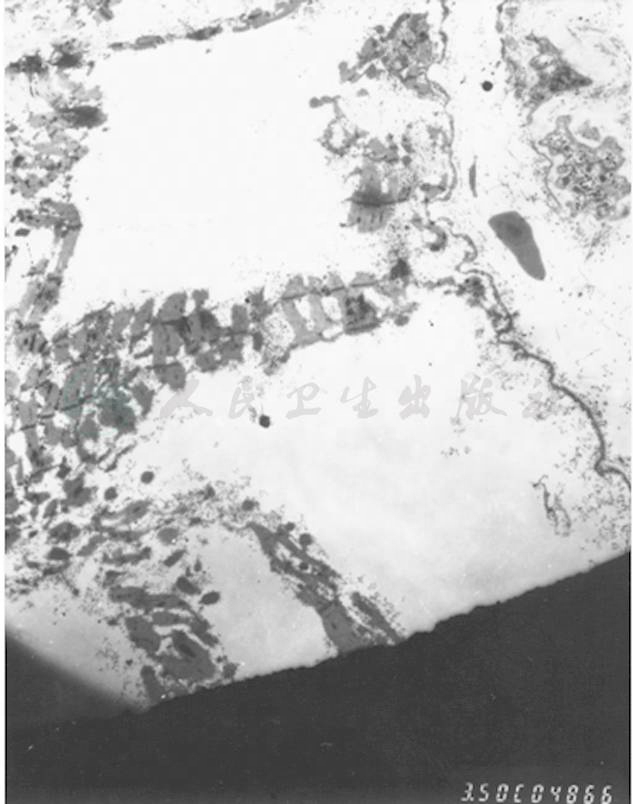
图5 肌活检(电镜)
肌纤维严重变性肿胀,肌丝结构模糊,部分肌纤维坏死,肌节结构消失,肌丝溶解,甚至整个肌纤维空泡化,仅存肌膜,呈空袋状。肌膜下、肌原纤维间可见大量糖原聚集(箭头所指)。细胞核多见固缩、变性
入院诊断
定位:肌肉。
定性:代谢性?变性?遗传性?
初步诊断:线粒体肌病?糖原累积病?
患者予以呼吸机维持呼吸、抗感染、护肝、营养支持等治疗,辅以肌苷、辅酶Q10、复合B族维生素等药物治疗,病情好转,顺利脱机出院。









