


 收藏
收藏 已收藏
已收藏脊膜膨出是一种病因不明但常见的神经系统发育畸形。脊柱在胚胎期发育缺陷,椎弓出现裂孔,形成脊柱裂,脊膜从裂隙膨出形成囊性肿物,即为脊膜膨出。如脊髓组织亦疝入囊肿,即为脊髓脊膜膨出。这些畸形可引起肢体瘫痪、大小便失禁或合并有其他畸形。
脊膜膨出的发生率约为1/5000~1/1000,各地区、种族间有一定差异。如脊柱裂在北爱尔兰发病率高达4.2‰,而亚洲仅为0.18‰~1.06‰。
一、胚胎学
胚胎14天后胚板背侧的外胚层中央部分细胞增生,局部增厚称为神经板。神经板继续向头端及尾端延伸,其两侧端逐渐隆起形成神经嵴。整个躯干部神经板两侧的神经嵴凹入成为神经沟,神经沟向深部发展,并互相对拢闭合发展成神经管(图1),神经管头端发展成脑,尾端发展成脊髓。
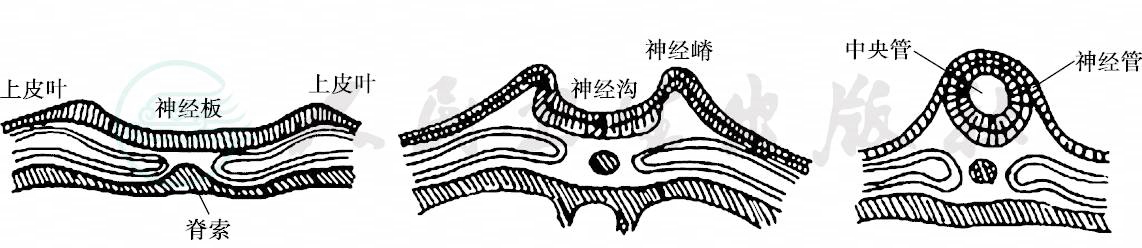
图1 胚胎神经管发育过程示意图
与外胚层神经板平行的中胚层索状结构称为脊索。脊索是脊柱的始基。随着神经管的纵行发育,脊索在胚胎体节的两侧伸出前、后突起。前突起发展成肋骨及横突;后突起发育成椎弓、椎板及棘突。整个脊索则发展为椎管,椎管围绕神经管。以上发育过程在胚胎第4周完成。脊柱裂是中胚层的发育障碍,导致椎管未完全闭合,棘突及椎板缺如或发育缺陷。椎弓有缺损后,脑脊液的压力作用使脊膜膨出超越皮肤平面,神经和脊髓也可向外膨出。腰部和腰骶部是脊柱裂最常见的部位。
二、病理分型
病理分型见图2。
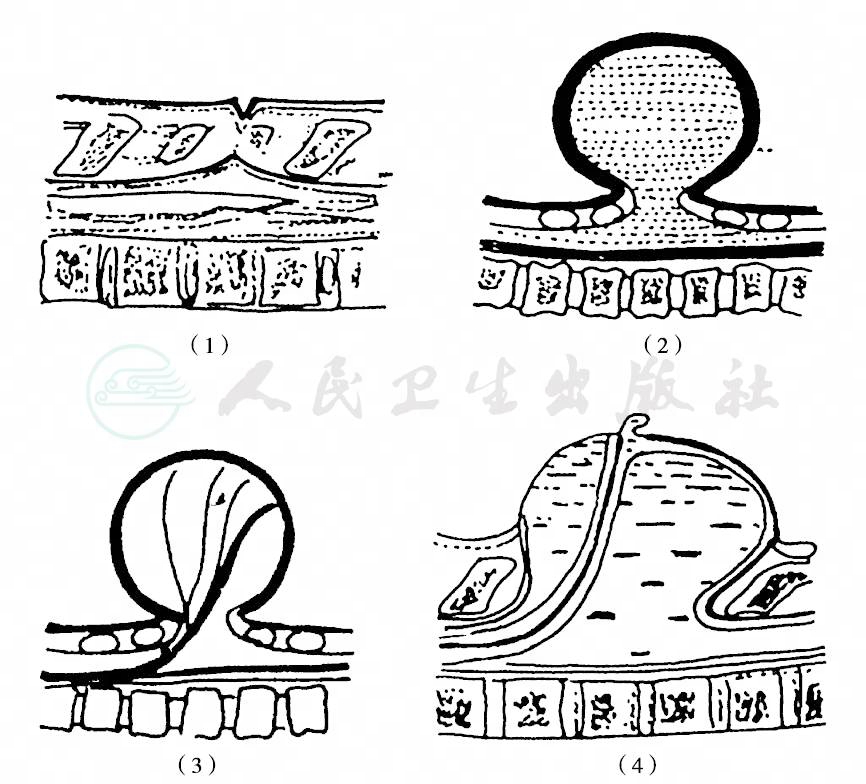
图2 脊膜膨出的病理分型
( 1)隐性脊柱裂; ( 2)脊膜膨出; ( 3)脊髓脊膜膨出; ( 4)脊髓外翻
隐性脊柱裂
多在L5~S1平面,局部椎弓缺损,脊柱呈现骨化障碍,但无脊膜或神经组织膨出。通常临床无明显症状。若神经根或终丝与裂孔处有纤维带粘连或压迫,可出现脊髓栓系综合征。患儿下肢乏力,感觉迟钝。随着年龄的增长,脊柱发育较快,神经的牵拉加重。隐性脊柱裂的皮肤表面时有毛发增生、色素沉着皮肤凹陷,有时合并皮样囊肿或脂肪瘤,称为潜毛窦。
脊膜膨出
神经管闭合完全,脊髓位置正常,囊壁由蛛网膜膜构成,囊外有皮肤覆盖,硬脊膜附着于棘突缺损边缘。脊膜膨出多位于腰骶部,囊腔内有脑脊液,有时有脊神经膨出黏附于囊壁上。表面皮肤正常或变薄。
脊髓脊膜膨出
表面有皮肤遮盖,有时中央很薄,膨出的囊腔内有脊髓或神经根存在。患儿多有神经功能障碍或截瘫。骶部的脊髓脊膜膨出有大小便失禁、足内翻、下垂或高弓足畸形;腰部脊髓脊膜膨出可出现双下肢麻痹、神经性膀胱、大便失禁等症状。
脊髓外翻
表面可见肉芽面,即裸露的脊髓,有时有溃疡,溃疡呈圆形或卵圆形。肉芽面周围有3~5mm宽的膜状组织,即为脊膜,其外围为正常皮肤,在皮肤的边缘深部可触得棘突的裂缘。脊髓顶部刚出生时平坦,以后略高出皮肤。
脊髓脊膜膨出还常合并有其他畸形,如阿-奇(Arnold-Chiari)畸形、畸胎瘤、皮样囊肿、骶骨发育不全、原发性泌尿系畸形等。
下面以典型病例探讨其诊断和治疗。
女性,4岁,生后发现腰骶部肿物伴大小便失禁生后至今。查体:臀部上方局限性隆起,中央凹陷,可见数根毛发,肛门括约肌松弛,鞍区针刺痛觉减退,耻骨上叩诊浊音。B超:膀胱残余尿量约500ml。
MRI示:腰骶脊柱裂,脊膜膨出,内容为脑脊液和神经组织,脊髓圆锥低位,脊髓栓系形成(图3)。
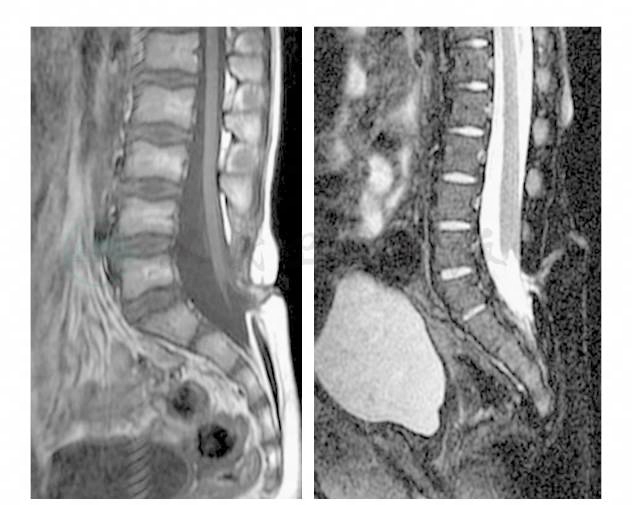
图3 腰骶脊柱裂,脊膜膨出脊髓圆锥低位,脊髓栓系形成
脊髓脊膜膨出手术可以保存神经组织,防止发育过程中神经继续受牵拉和压迫,以免畸形不断发展,加剧神经的功能障碍。但已有神经肌肉功能缺损者,手术不能使其恢复。一般认为病儿有轻度的下肢瘫痪和大小便失禁时,仍应早期手术,术后再做功能重建及括约肌成形术。出生后早期手术感染危险少,神经根受到牵拉和压迫较轻,其支配区肌肉功能损害较小。
本例患者手术将突出于椎管外的脊髓和神经样结构还纳,终丝切断、栓系松解,膨出的硬脊膜翻转缝合,在椎管内水平重建硬脊膜腔,逐层翻转、加固缝合,皮肤塑性缝合。









